


1. Introduction
Polymer electrolyte fuel cells (PEFCs), which convert chemical energy directly into electricity through electrochemical reactions, are considered to be ideal energy conversion devices for future portable and stationary applications owing to their high efficiency and environmentally benign nature [1]. Although PEFC technologies are currently approaching commercialization, their high cost and insufficient long-term durability remain as major bottlenecks that hinder their widespread deployment. The high cost of PEFCs stems largely from the use of Pt-based electrocatalysts for both electrode reactions: hydrogen oxidation reaction (HOR) at the anode and oxygen reduction reaction (ORR) at the cathode. Particularly, large amounts of Pt are required at the cathode of PEFCs in order to circumvent the intrinsically sluggish kinetics of the ORR [2-4]. Hence, to make PEFCs more economically viable, tremendous effort has been devoted to developing highly active, durable, and low-cost ORR catalysts. Pt−M (M=transition metal: Fe, Co, Ni, etc) alloys and M@Pt core−shell-type catalysts have been at the forefront of this research effort [3,4]. Moreover, the genesis of shape and morphology-controlled nanoparticles (NPs) of alloy compositions further enhanced the ORR activity of Pt-based ORR catalysts. Another major direction is based on non-precious metal catalysts (NPMCs). Notable classes of NPMCs include heteroatom (e.g. N, S, B, and P)-doped carbon [5-10], metal oxide or metal oxide-carbon composites [11-15], and transition metal and nitrogen codoped carbon (M−N/C) catalysts [16-40].
Among NPMCs, the M−N/C catalysts have received particular interest owing to their high ORR activity compared to those of other classes of NPMCs. Jasinski first demonstrated that cobalt phthalocyanine (Pc) molecules can catalyze ORR in alkaline media. Early research efforts in M−N/C catalysts were focused mainly on metallomacrocycle-based molecular electrocatalysis [41-43]. A significant advance in M−N/C catalysts was achieved by Jahnke et al. who demonstrated that heat-treatment of metallomacrocycles at high temperatures significantly improves both the activity and stability of the catalyst for ORR [44]. Yeager and co-workers subsequently showed that heat-treatment of a mixture containing a metal precursor, N-containing polymer, and carbon support could also produce ORR catalysts with activities comparable to those of pyrolyzed macrocycle-based catalysts [45].
On account of these advances, high-temperature pyrolysis has been generally regarded as an essential step in the preparation of high-performance M−N/C catalysts. The pyrolysis step imparts high electrical conductivity and structural integrity in final M−N/C catalysts. Nevertheless, this step inevitably generates heterogeneity in the metal sites of resulting catalysts, thereby making the identification of active species challenging. Therefore, the enhancement in activity has been achieved mainly via experimental optimizations of precursors, pyrolysis temperatures, and post-synthetic treatments.
Development of an efficient M−N/C catalyst can be accelerated by advancing our understanding of the active sites. To date, it has been established that there are two major metal species in pyrolyzed M−N/C catalysts: (i) atomically dispersed metal species stabilized by coordination bonds with N atoms in carbon lattice (M−Nx sites) and ii) inorganic NPs (metals, metal carbides, metal nitrides, or metal sulfides depending on the type of precursors employed) [46-49]. Detailed insights into the nature of active sites have been developed through a variety of spectroscopic methods. Fe−N/C catalysts have been studied most widely owing to their superior ORR activity when compared to other M−N/C catalysts and the availability of 57Fe Mössbauer spectroscopy that enables detailed investigation of Fe-related sites. For pyrolyzed Fe–N/C catalysts, there is a growing consensus that the ORR activity originates primarily from Fe−Nx species [46,47]. However, inorganic particles composed of metallic iron and iron carbide have recently been suggested as a new type of active species [48,49].
In this focus review, we first summarize the recent advances in spectroscopic and electrochemical methods for the identification of Fe−Nx sites in pyrolyzed Fe−N/C catalysts, and their structure-activity relationship for the ORR. We introduce synthetic strategies for the fabrication of high-performance pyrolyzed Fe−N/C catalysts with higher density of active sites. We discuss some examples of synergistic enhancement of ORR activity in bimetallic FeM−N/C catalysts. Next, we present recent findings on new potential active species, namely iron or iron carbide encapsulated with graphitic carbon shells. Finally, we summarize this review and provide an outlook for this field.
2. Characterization of Fe−Nx Sites by Spectroscopy and Electrochemistry
Significant effort has been devoted to identification of chemical states, local structures, and catalytic roles of atomically dispersed Fe−Nx sites in pyrolyzed Fe−N/C catalysts. Dodelet and co-workers attempted to analyze surfaces of pyrolyzed Fe−N/C catalysts using time of flight secondary ion mass spectroscopy (TOF-SIMS) [50]. The TOF-SIMS spectrum of an Fe−N/C catalyst pyrolyzed at temperatures above 600 °C (Fig. 1(a)) showed a signature corresponding to FeC2N4+ ion. The analysis of TOF-SIMS data revealed a good correlation between ORR activity and the number of such ions detected, thus suggesting that the active sites are comprised of Fe ion bonded to two N atoms in a phenanthroline-like carbon framework [50]. Chen et al. later applied TOF-SIMS-based ion imaging to Fe−N/C catalysts, providing an insight into the chemical properties of active sites where CxNy species are adjacent to an Fe center [51]. X-ray photoelectron spectroscopy (XPS) is a widely used tool for uncovering the existence of Fe−Nx sites. The deconvoluted peak in N 1s XPS spectrum at 399.5−399.8 eV, located between the peaks arising from pyridinic and pyrrolic nitrogens (Fig. 1(b)), can be typically assigned as Fe−Nx site [52]. Density functional theory (DFT) calculations revealed that the peak for pyridinic N shifts to a higher binding energy when coordinated with Fe, thus corroborating the experimental results [53].
X-ray absorption spectroscopy (XAS) is an element-specific technique applicable to non-crystalline materials, and is a highly versatile tool for the characterization of Fe−N/C catalysts [37,40]. Jaouen et al. prepared Fe−N/C catalysts containing only Fe−Nx sites, and analyzed the catalysts using X-ray absorption near-edge structure (XANES) spectroscopy [54]. By combining experimental XANES data with XANES simulation study and DFT calculations, the active sites in their Fe−N/C catalysts were found to consist of a FeN4C12 moiety with a slightly off-center Fe atom (Fig. 1(c)). Extended X-ray absorption fine structure (EXAFS) is the oscillatory response that appears in the higher photon energy region of XAS spectra, and can provide information about the coordinating atom, coordination number, and inter-atomic distances around the X-ray absorbing atom (Fe in this case). The majority of pyrolyzed Fe−N/C catalysts show a major peak in a radial distribution function from EXAFS spectra at 1.6−1.8 Å that corresponds to Fe−N/O bonds [55]. An additional peak at 2.0−2.4 Å indicates the presence of Fe-based particles such as metallic Fe and Fe3C, formed by hightemperature pyrolysis above 700 °C (discussion of such particles is provided in Section 6). We note that XAS is a bulk-sensitive analytical technique and therefore can provide only average structural information to be obtained. For this reason, XAS data must be interpreted with great caution; otherwise it can lead to misleading conclusions.
57Fe Mössbauer spectroscopy is perhaps the most powerful tool that can distinguish different Fe-based species owing to its high sensitivity to coordination environment-induced changes in electronic and magnetic states. The most important advantage of the Mössbauer analysis over other techniques is its ability to differentiate Fe−Nx sites with similar local structure but different oxidation and spin states (Fig. 1(d)) [46,47,56].
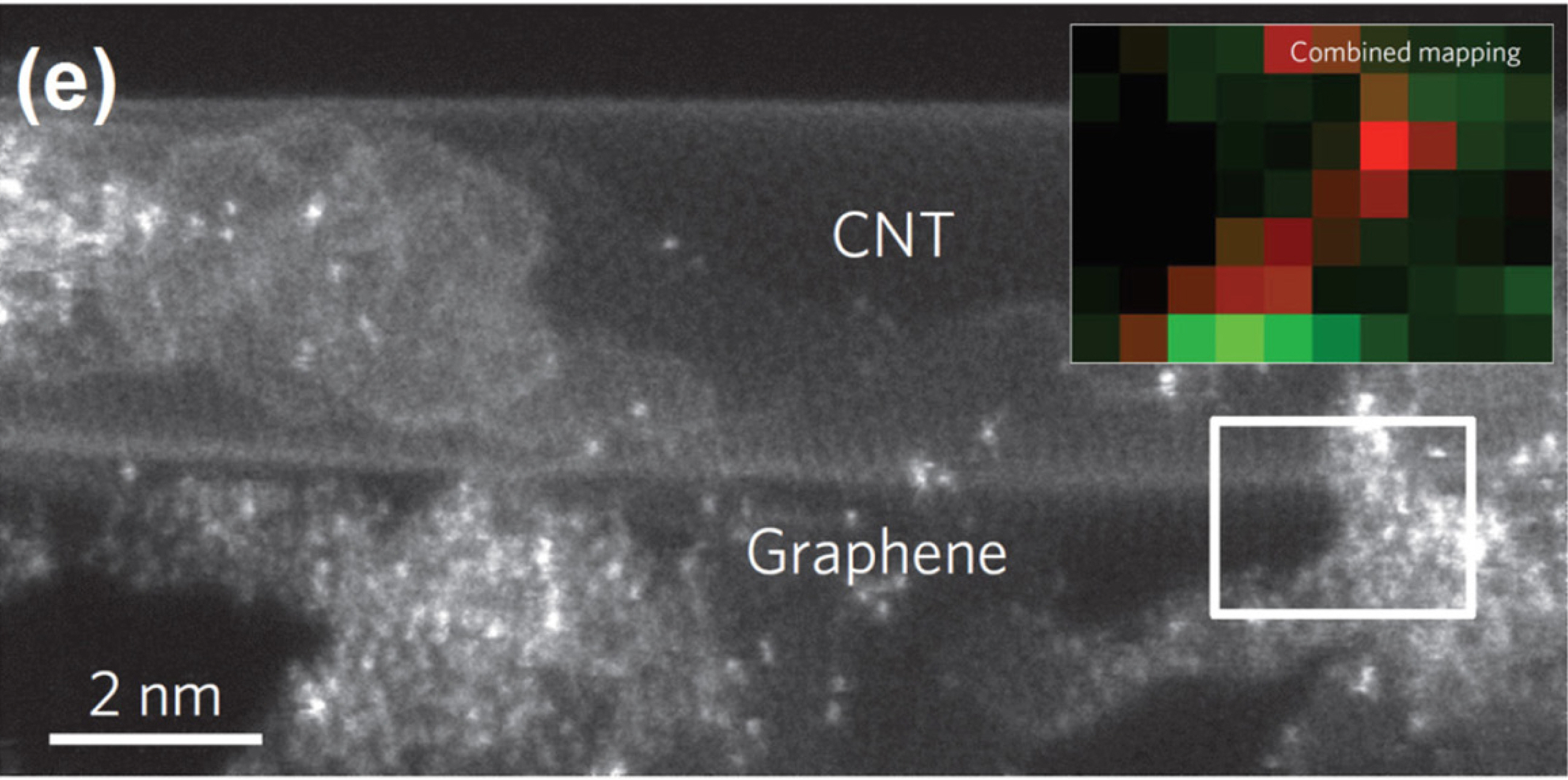
Complementary to the above spectroscopic methods, transmission electron microscopy (TEM) is an important tool in characterizing pyrolyzed Fe−N/C catalysts. The recent advances in TEM allow direct observation of a single atomic Fe site (considered as Fe−Nx site). Using aberration-corrected scanning TEM, Dai et al. observed the dispersion of individual Fe atoms in their Fe−N/C catalyst, and collected elemental mapping data that showed that Fe and N atoms are present in close proximity, thus providing direct evidence for the existence of Fe−Nx species (Fig. 1(e)) [57].
Fig. 1.
(a) TOF-SIMS spectrum of a pyrolyzed Fe−N/C catalyst at a selected mass range. Reprinted with permission from Ref. [50].
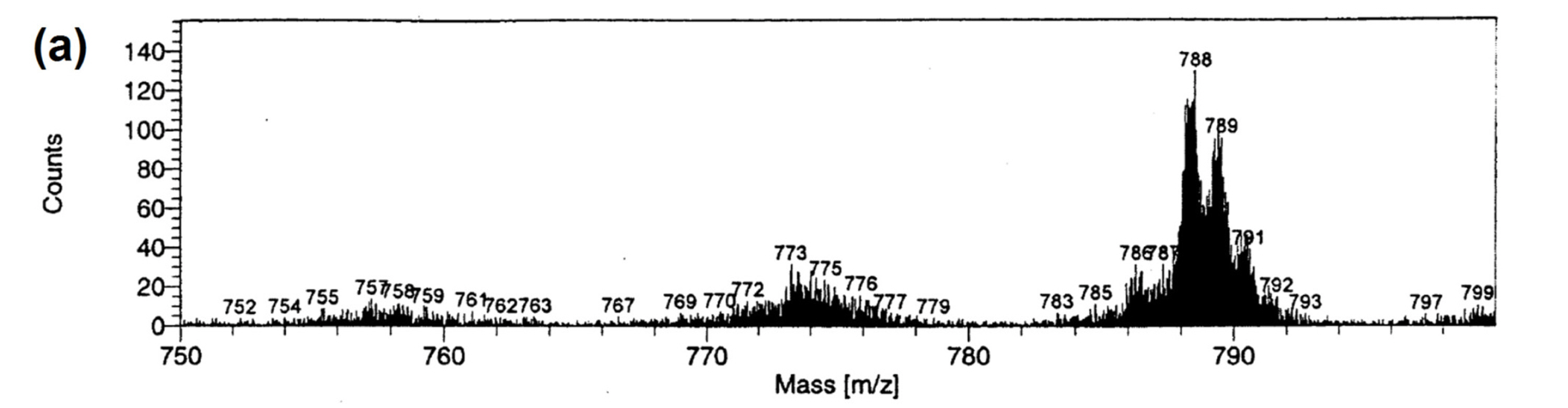
(b) Nitrogen 1s XPS spectrum of a pyrolyzed Fe−N/C catalyst. Reprinted with permission from Ref. [52].
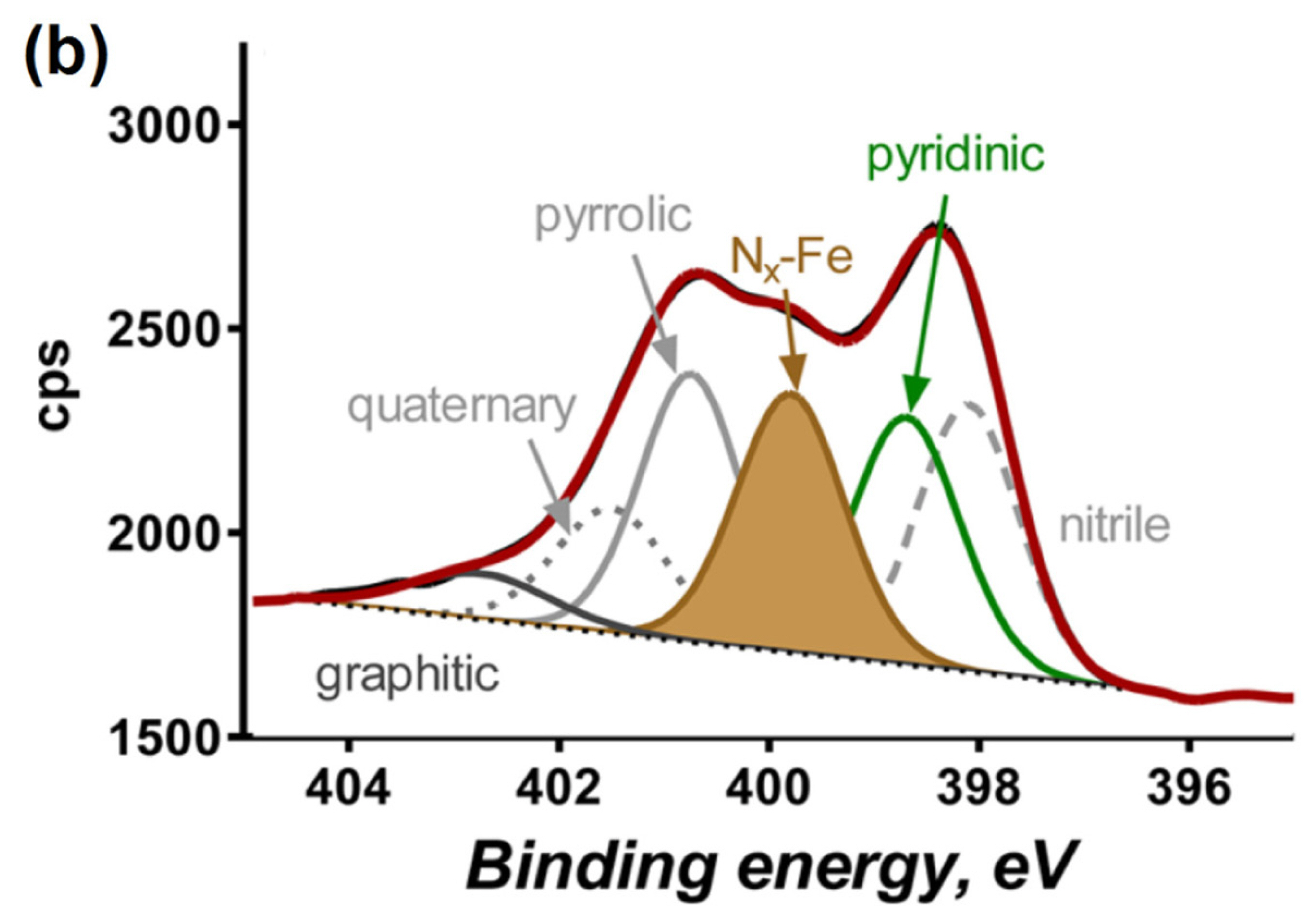
(c) XANES spectrum of Fe0.5 catalyst (black dashed line) and a simulated spectrum (red line). Orange, blue, gray, and red spheres indicate iron, nitrogen, carbon, and oxygen atoms, respectively. Reprinted with permission from Ref. [54].
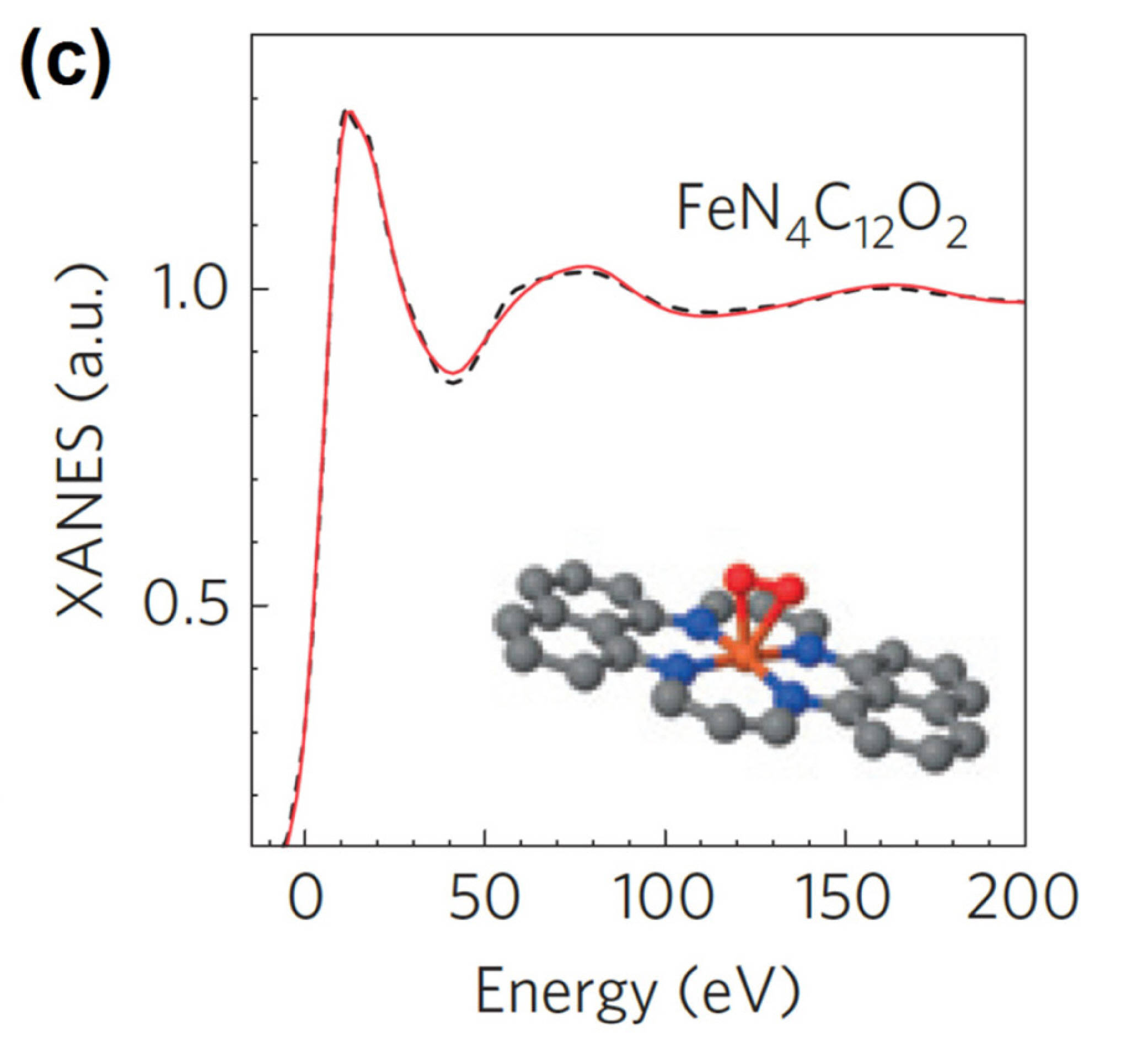
(d) 57Fe Mössbauer spectrum with deconvoluted peaks corresponding to identified Fe species. Reprinted with permission from Ref. [47].
(e) Aberration-corrected annular dark-field transmission electron micrograph of NT-G catalyst. Inset of (e) shows the elemental mapping image of NT-G catalyst. Red and green squares represent the signals from Fe and N, respectively. Reprinted with permission from Ref. [57].
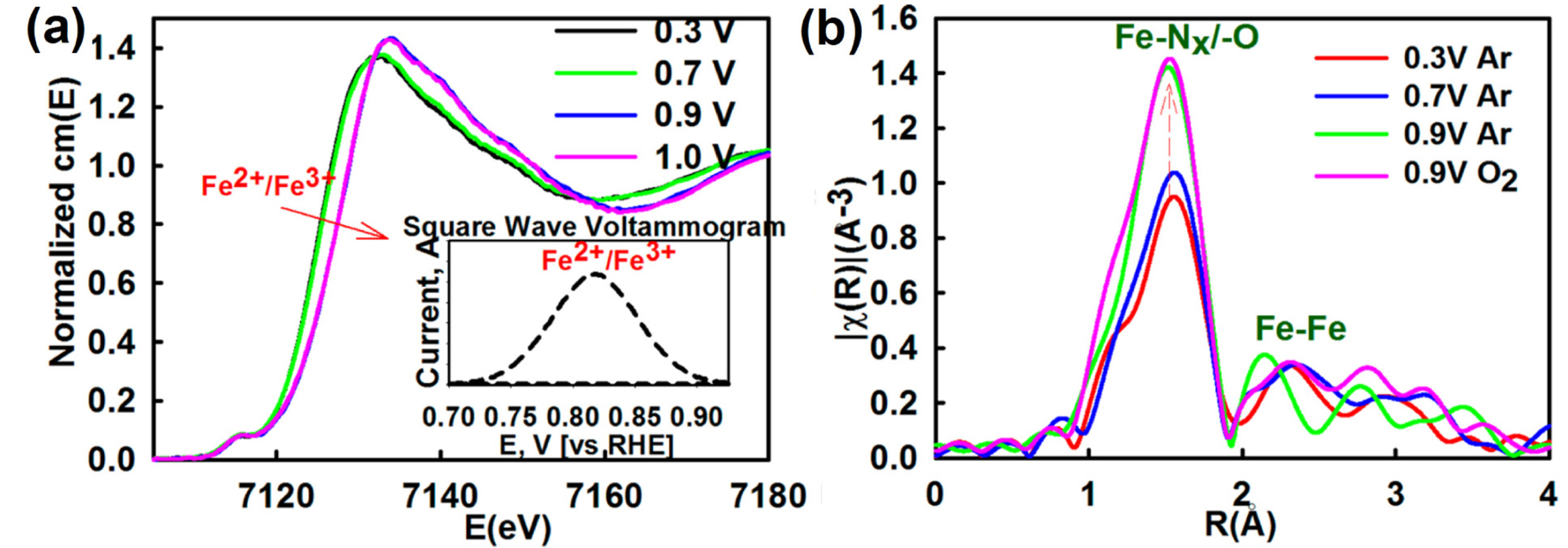
Although the catalytic role of Fe−Nx sites in the ORR has not yet been fully clarified, some spectroscopic and electrochemical studies have provided evidence for the direct participation of Fe−Nx sites in the ORR. Mukerjee et al. have exploited electro-chemical in-situ XAS to investigate the chemical and structural changes in the Fe−Nx species during the ORR [55,58]. Their studies, where XAS was performed under controlled potentials in N2- or O2-saturated saturated electrolyte, showed three significant observations: (i) Fe2+/Fe3+ redox reactions occur around the potential at which the ORR starts (Fig. 2(a)); (ii) Fe−N/O coordination number (interatomic distance of Fe−N and Fe−O is hardly distinguishable by EXAFS) increases as the applied potential increases (Fig. 2(b)); and (iii) the relationship observed between OH coverage and potential matched the ORR polarization curves (Fig. 2(c)). Observations (i) and (ii) are indicative of an OH adsorption process (Eq. (1)) by Fe2+-oxidation and concerted water activation [55,58];
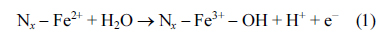
Fig. 2.
(a) Electrochemical Fe K-edge in situ XANES spectra and (b) Fourier-transformed EXAFS spectra of PVAG-Fe catalyst measured in 0.1 M HClO4. Inset of (a) shows square wave voltammogram showing Fe2+ → Fe3+ oxidation peak. Reprinted with permission from Ref. [55].
(c) OH coverage estimated from electrochemical in-situ EXAFS analysis for a set of pyrolyzed Fe−N/C catalysts (indicated by symbols and lines), and a simulated curve (dashed lines). Reprinted with permission from Ref. [58].
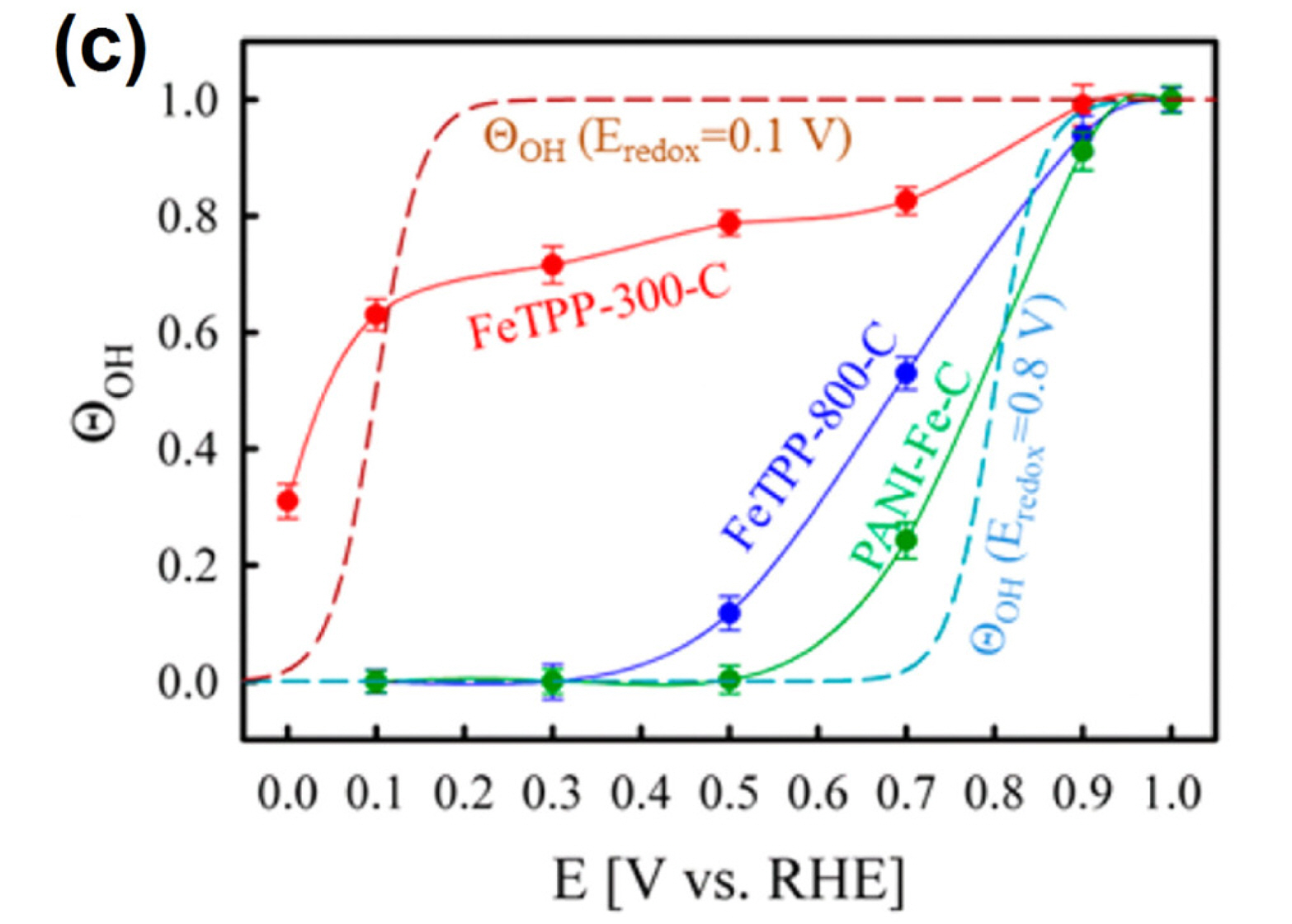
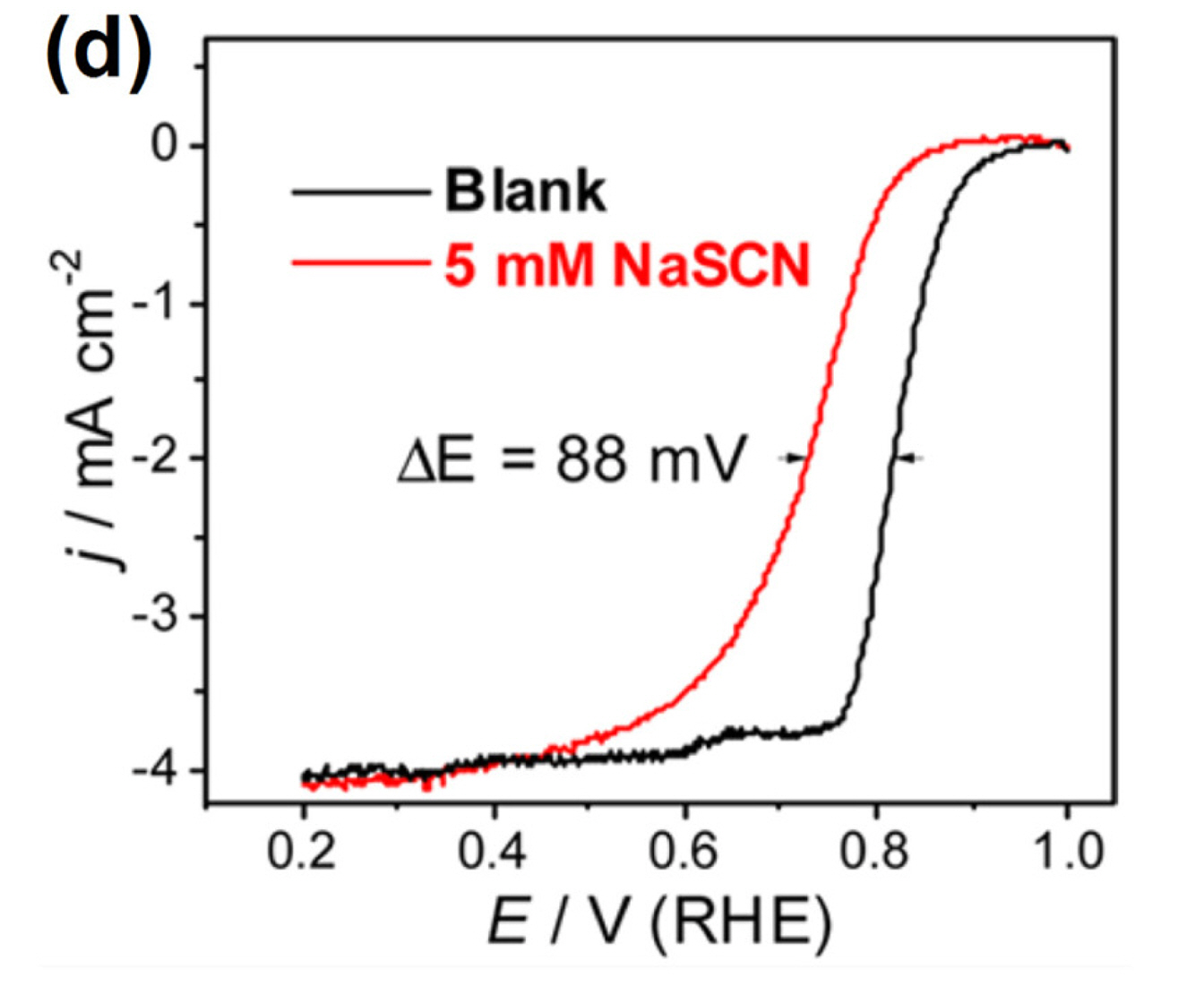
Observation (iii) can be corroborated by the active site blocking effect exerted by the OH adsorbate. In this case, the reaction rate was governed by the potential-dependent desorption of OH species accompanied by the reduction of Fe3+ to Fe2+, which generates Fe2+−Nx species that allow subsequent adsorption and reduction of O2 to occur. Therefore, the active species was suggested to be Fe2+−Nx [58].
Another method for identifying the involvement of Fe−Nx sites in ORR utilizes probe molecules such as CN−, SCN−, and H2S (Fig. 2(d)) [59-61]. When these probe molecules are introduced in the electrolyte, the ORR activity of Fe−N/C catalysts generally declines as a result of strong adsorption of probe molecules on Fe−Nx sites, which prevents adsorption of O2. Such poisoning experiments are simple, and thus used widely to investigate the catalytic role of Fe−Nx species in pyrolyzed Fe−N/C catalysts.
Precise quantification of Fe−Nx site density is of particular importance for the evaluation of intrinsic activity of developed Fe−N/C catalysts, fair comparison of activity between the catalysts from different groups, and the development of improved synthetic methods for the preparation of novel Fe−N/C catalysts with higher density of Fe−Nx sites. The chemical probe method has sometimes been exploited for determining the number of Fe−Nx sites. Strasser and co-workers analyzed the Fe−Nx site density using a CO chemisorption method [62]. Due to the weak interactions between Fe−Nx sites and CO molecules at a room temperature, they performed CO pulse chemisorption at 193 K in conjunction with temperature-programmed desorption. The density of Fe−Nx sites was calculated using the quantity of desorbed CO molecules. More recently, Kucernak et al. employed nitrite anion (NO2−) as a probe molecule [63]. Without an applied potential, the Fe−Nx sites are easily poisoned in NO2− solution. During electrochemical stripping, adsorbed nitrite is reduced through a five-electron transfer per Fe−Nx site, thereby enabling quantification of Fe−Nx sites. However, nitrite stripping is only possible at pH 5.2, where the nitrite anion is stable, whereas the majority of ORR activity measurements are conducted under strongly acidic or alkaline conditions.
3. ORR Activity-Structure Correlation
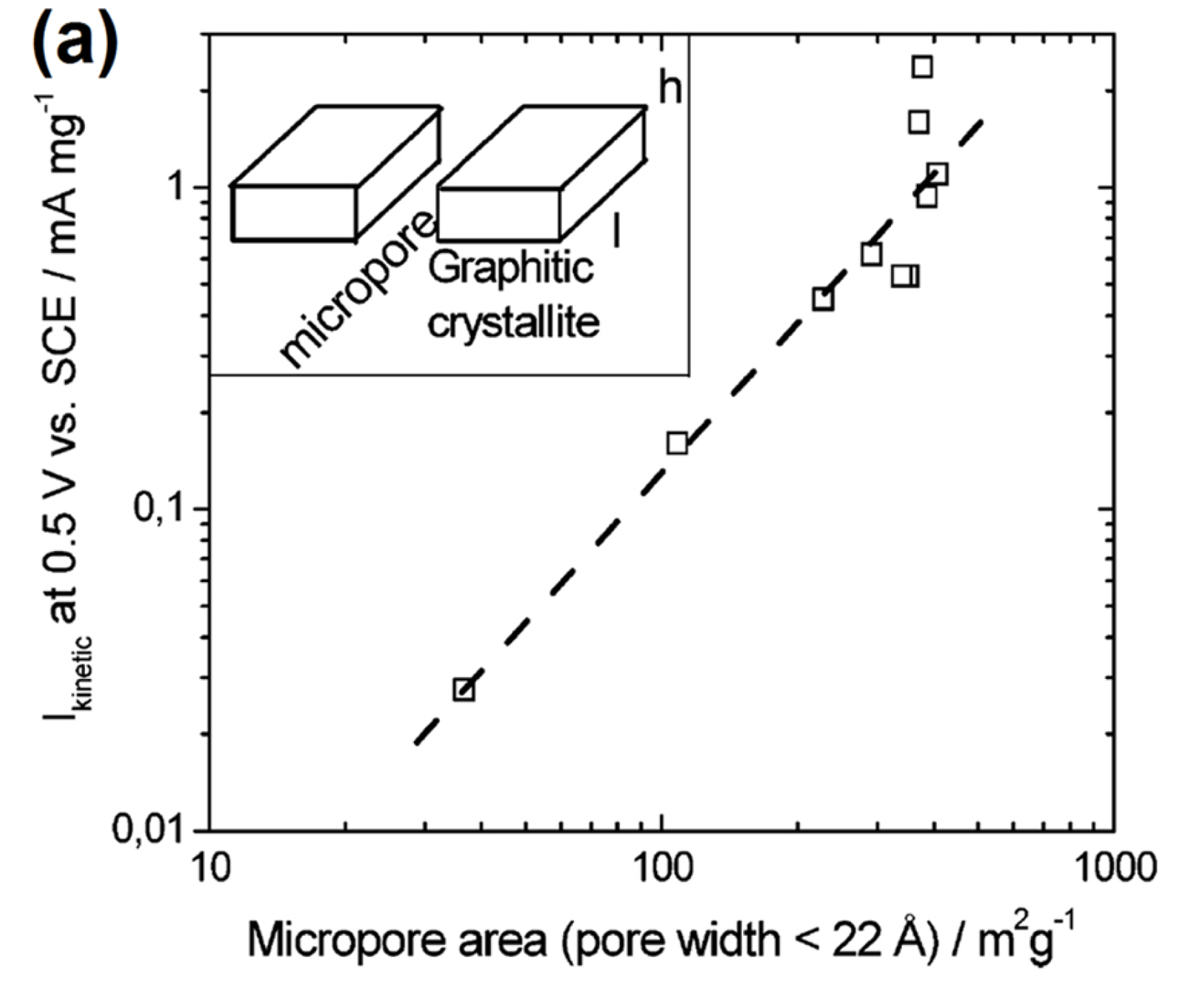
Establishing the relationship between the catalyst structure and ORR activity is of utmost importance for the rational design of advanced Fe−N/C catalysts. To date, several structural parameters have been identified to exert strong influences on the ORR activity of Fe−N/C catalysts. Dodelet et al. identified that the micropore surface area of carbon supports play an important role in dictating ORR activity. They prepared a set of Fe−N/C catalysts by heat-treatment of iron(II) acetate-impregnated carbon black in NH3 for 5 to 50 min [64]. They postulated that NH3-treatment activates the catalyst via formation of micropores, in which Fe and NH3 react to generate Fe−Nx sites. As the time of NH3-treatment increased, the catalysts became increasingly microporous with increased micropore surface area (pore size < 2.2 nm). The ORR activity of the catalysts in acidic media correlated well with the micropore surface area, thus indicating that the active sites are located in the micropores (Fig. 3(a)).
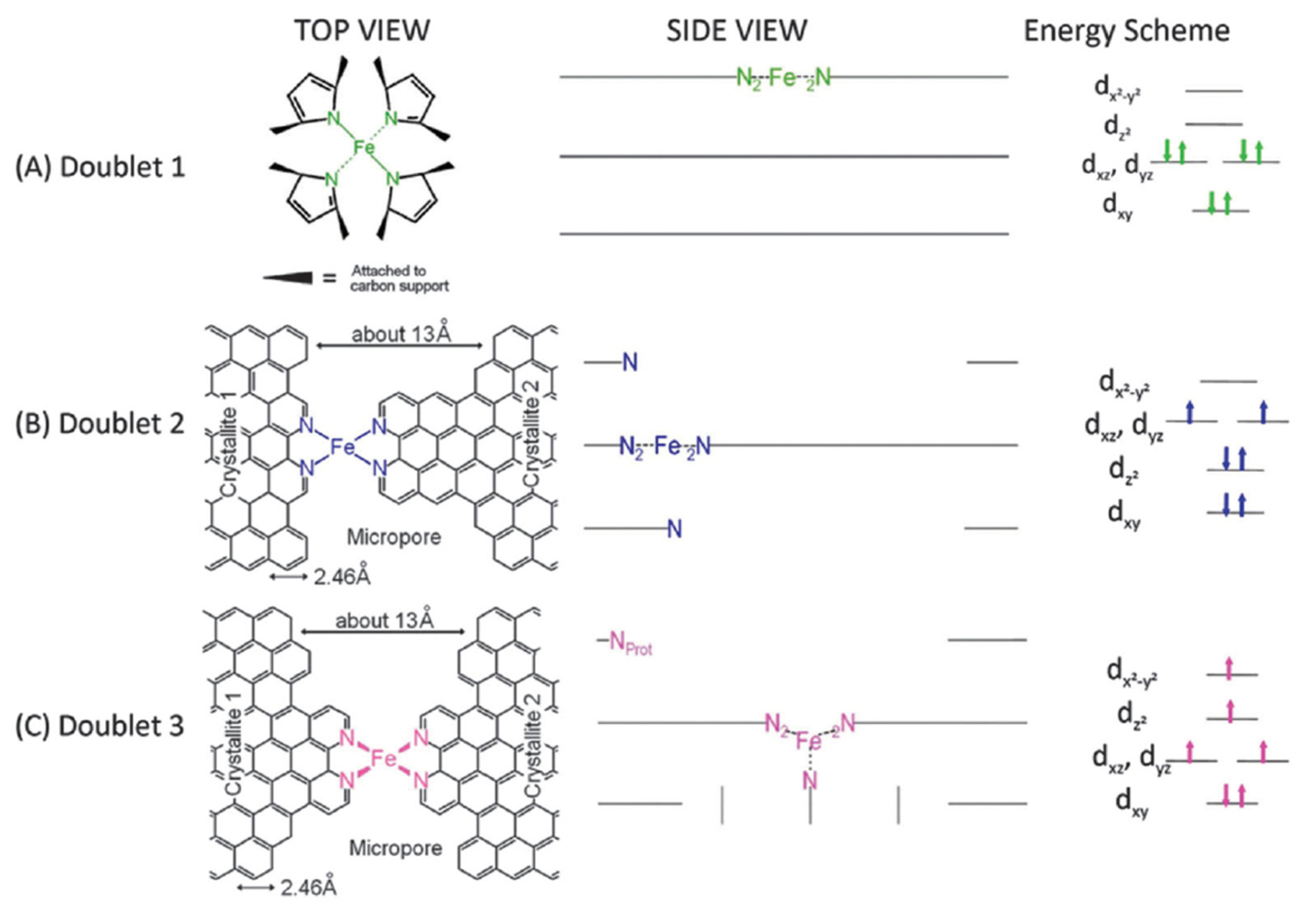
As described in Section 2, Mössbauer spectroscopy can provide the most detailed information about the geometric and electronic structures of Fe−N/C catalysts. Several types of Fe−Nx sites with different electronic structures have been identified based on their nuclear interaction parameters, namely isomer shift and quadrupole splitting. Fe−Nx sites exhibit a doublet Mössbauer signature, and are therefore labeled as the D (doublet) sites. D1 site is assigned to in-plain low-spin (S = 0) ferrous Fe−N4 site, D2 site is crystal-line FePc-like Fe−N4 site with pseudo-octahedral coordination with axial N atoms from the adjacent FePc, and D3 site comprises the Fe center that is coordinated with five N atoms, and the Fe2+ is positioned out-of-plane, in a direction towards the axially- positioned N atom (Fig. 4). We note that the D2 site contributes to the ORR activity negligibly, due to its coordination environment and fully occupied dz2 orbital of Fe center. Dodelet and co-workers found that, among the three D sites, D1 site was found to be the major active site in pyrolyzed iron porphyrin catalysts for ORR in acidic media (Fig. 3(b)) with an average turnover frequency (TOF) of ~0.01−1 s−1 at 0.8 V (vs reversible hydrogen electrode; RHE) [46,47,65,66]. The same group later suggested that NH3-treated Fe−N/C catalysts possessed another active site, D3. However, in-depth investigation of D3 site and its relation to the ORR activity has not yet been widely accepted [47,67].
Fig. 3.
(a) Correlation between the micropore area and kinetic current at 0.5 V (vs saturated calomel electrode; SCE) of NH3-treated Fe−N/C catalysts. Reprinted with permission from Ref. [64].
(b) Correlation between the mass activity for the direct oxygen reduction to H2O (4-electron ORR) and the amount of D1 (in-plane Fe−N4) species. Reprinted with permission from Ref. [46].
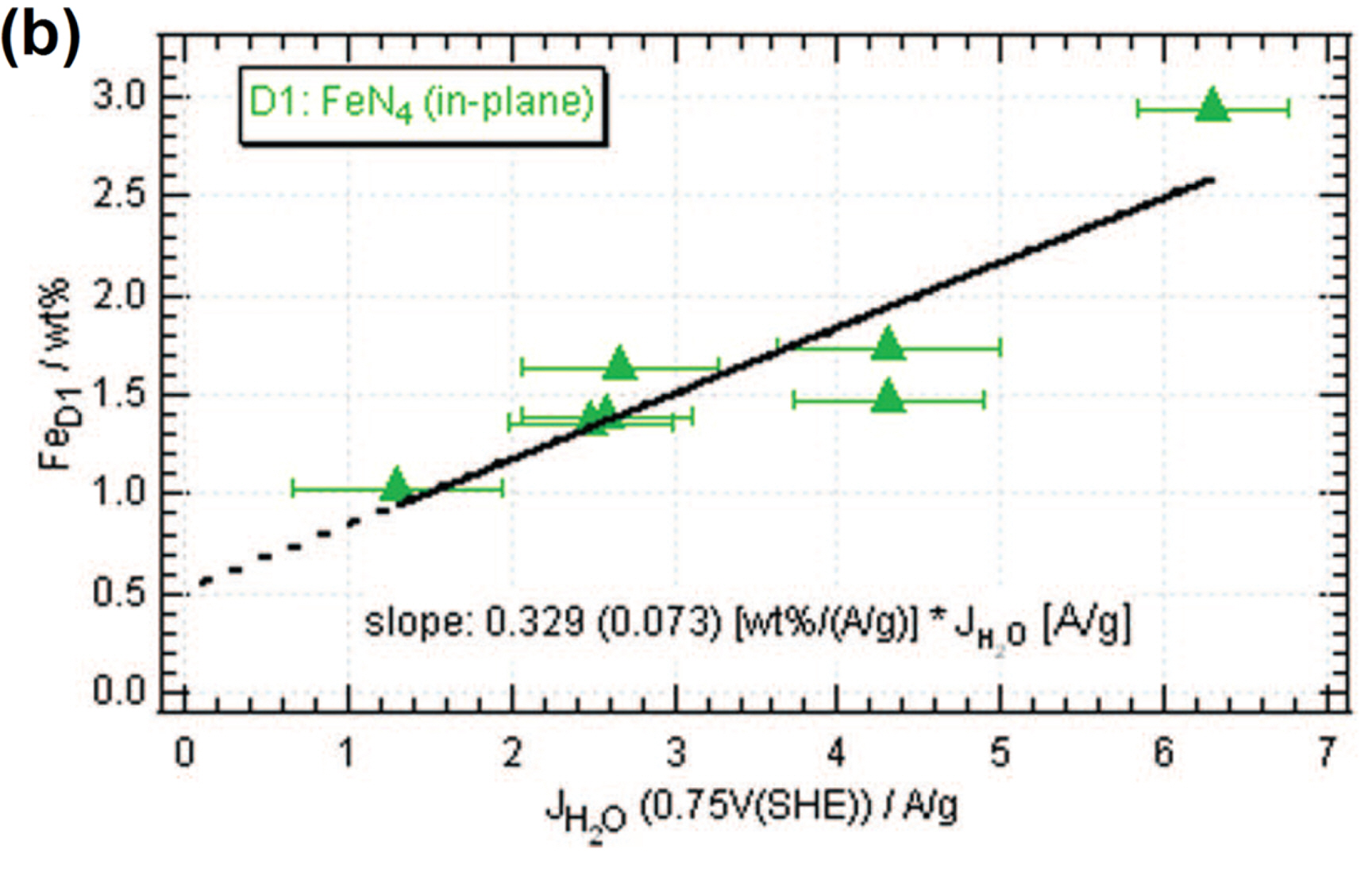
Fig. 4.
Geometric and electronic structures of D1, D2, and D3 Fe−Nx species. Reprinted with permission from Ref. [47].
4. Design of Fe−N/C Catalysts with Abundant Fe−Nx Sites
As discussed in earlier sections, investigations on the active sites in Fe−N/C catalysts over the past decades have provided a plausible evidence that established Fe−Nx sites as the main active species of pyrolyzed Fe−N/C catalysts. However, high-temperature pyrolysis usually results in aggregation of Fe atoms and encapsulation of the resulting particles with graphitic carbon shells. Inorganic particles have been generally regarded as impurities, i.e., species that diminish the ORR activity. Post acid-treatment and heat-treatment under corrosive NH3 atmosphere were exploited to remove the impurity particles in order to boost the ORR activity [24-28,31,32,34, 36,37,40,47,54,57-59,62-67]. However, rational methods for selective creation of active Fe−Nx species during pyrolysis, while minimizing the formation of inactive particles, are still lacking.
Joo and co-workers have recently developed a general ‘silica-protective-layer-assisted’ strategy that can preferentially generate active Fe−Nx sites, leading to high-performance Fe−N/C catalysts (Fig. 5(a)) [68]. In this method, porphyrin-coated carbon nanotubes (CNTs) are overcoated with a silica layer, which is followed by high-temperature pyrolysis and silica etching to give CNTs coated with a thin layer of porphyrinic carbon (CNT/PC). 57Fe Mössbauer spectroscopy, X-ray diffraction (XRD), high-resolution TEM, and XAS analyses revealed consistently that CNT/PC possessed a higher density of Fe−Nx sites than the CNT/PC prepared without the silica coating (CNT/PC_w/o SiO2), which contained instead a significant quantity of large Fe and Fe3C particles (Fig. 5(b) and 5(c)). Temperature-controlled in-situ XAS suggested the presence of interactions between the silica and the Fe−N4 moiety in the porphyrin precursor, which may stabilize the Fe−Nx sites during the high-temperature pyrolysis. The CNT/PC with a higher number of active Fe−Nx sites exhibited 3-4 times higher ORR activity than CNT/PC_w/o SiO2 in both acidic and alkaline electrolytes (Fig. 5(d) and 5(e)). Importantly, a CNT/PC-based cathode exhibited very high current and power densities in an alkaline anion exchange membrane fuel cell (AEMFC) when compared to values of other NPMCs. In addition, a PEMFC with a CNT/PC-based cathode exhibited a high volumetric current density of 320 A cm−3, which is comparable to the US DOE 2020 target (300 A cm−3).
Fig. 5.
(a) Schematic illustration of the synthetic procedure employed for the fabrication of a CNT/PC catalyst. 57Fe Mössbauer spectra of (b) CNT/PC and (c) CNT/PC_w/o SiO2. ORR polarization curves for CNT/PC and CNT/PC_w/o SiO2 measured in (d) 0.1 M KOH and (e) 0.1 M HClO4. Insets in (d) and (e) represent the kinetic current densities of the catalysts calculated at 0.9 and 0.8 V, respectively. Reprinted with permission from Ref. [68].

The addition of elemental sulfur to the precursor mixture was reported as an alternative approach to preventing the destruction of Fe−Nx sites [69,70]. Kramm and co-workers performed a thermogravimetry coupled mass spectrometry (TGA-MS) study, which revealed that the destruction of active sites in sulfur-free porphyrin precursor occurred in two temperature regions, namely 450−520 °C and 550−650 °C, whereas no collapse of active sites was observed even at the higher temperature range for the sulfur-porphyrin mixture [69]. On the basis of high-temperature XRD results, they hypothesized that the second destruction temperature range is related to the reduction of intermediate iron oxides by carbon, and their subsequent transformation into Fe3C. During pyrolysis in the presence of sulfur, iron compounds can react with sulfur, thus giving rise to iron sulfide instead of Fe3C (Fig. 6(a)). The ORR activity of the sulfur-containing Fe−N/C catalyst was about 10 times higher than that of the sulfur-free catalyst (Fig. 6(b)). This increase in activity can be explained by the increased density of Fe−Nx active sites. The promoting effect of sulfur on catalyst activity was also observed for the Zelenay group’s catalysts derived from polyaniline (PANI), where (NH4)2S2O8 was used as an oxidative initiator for aniline polymerization. The (NH4)2S2O8-containing catalyst possessed a lower quantity of Fe3C species as evidenced by XANES analysis, and exhibited enhanced ORR activity [71].
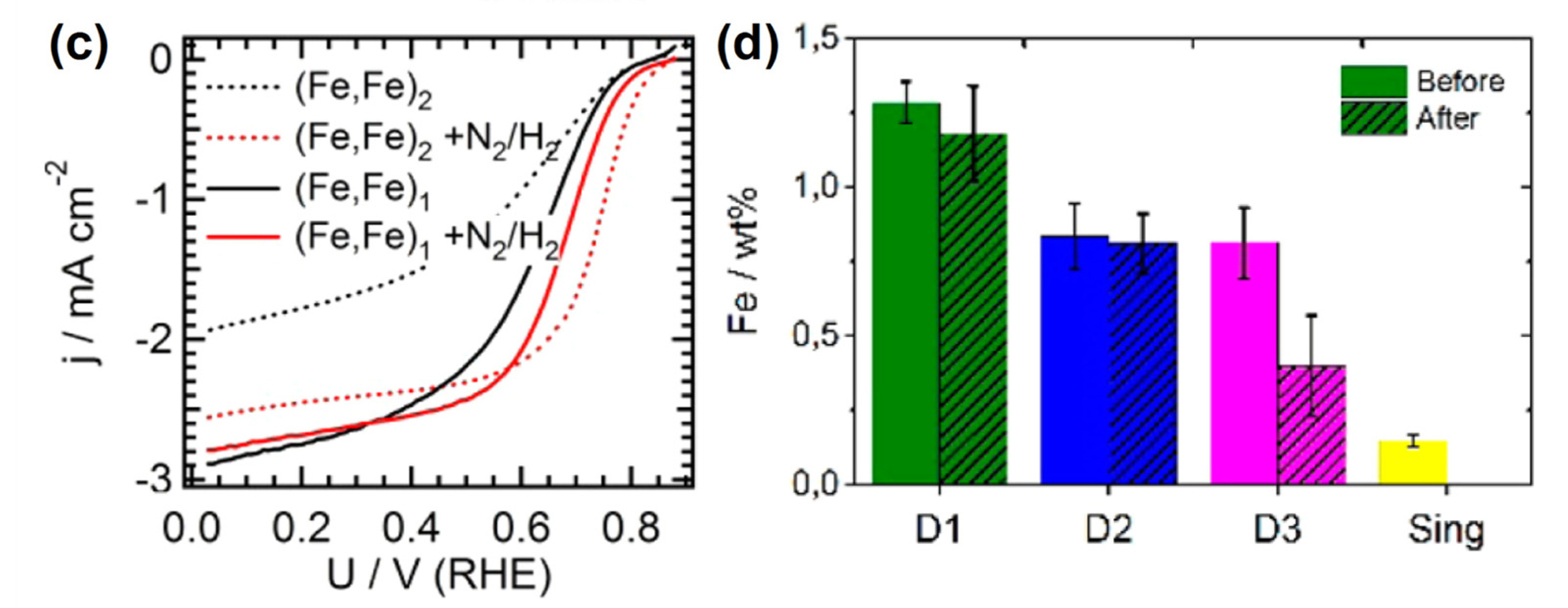
Kramm et al. suggested that Fe-based inorganic particles (Fe and Fe3C) can be removed by secondary heat-treatment under diluted H2 gas conditions [66]. Indeed, the ORR activity of a pyrolyzed porphyrin catalyst was improved after the second heat-treatment under 10% H2/N2 at 800 °C, followed by acid leaching (Fig. 6(c)). Mössbauer spectroscopy showed that the Fe particles were removed from the initial catalyst by the H2-treatment, thus yielding Fe−N/C catalyst incorporating only Fe−Nx sites (Fig. 6(d)).
Fig. 6.
(a) X-ray diffraction patterns and (b) ORR Tafel plots for heat-treated iron porphyrin catalysts. ‘+S’ and ‘−S’ indicate that the catalyst was pyrolyzed with and without elemental sulfur, respectively. Reprinted with permission from Ref. [69].

(c) ORR polarization curves and (d) the quantities of D1, D2, D3, and singlet species determined by 57Fe Mössbauer spectroscopy for heat-treated iron porphyrin catalysts before and after H2-purification process. Reprinted with permission from Ref. [66].
5. Bimetallic Synergism in M−N/C Catalysts
Cytochrome c oxidase (CcO) is an enzyme that catalyzes ORR at the end of the respiratory cycle in biological systems [72]. CcO is comprised of a heme Fe2+ with an axial imidazolic N and a non-heme Cu atom at a distance of ~4.5 Å from Fe [73]. The catalytic center shows a very high TOF and a perfect selectivity for H2O (4-electron ORR). The distal Cu atom has been shown to assist in the electron transfer to oxygen and the cleavage of O−O bond [74-76]. Such active sites with a sophisticated binuclear structure have inspired scientists to attempt and reproduce analogous structure in heterogeneous pyrolyzed Fe−N/C catalysts.
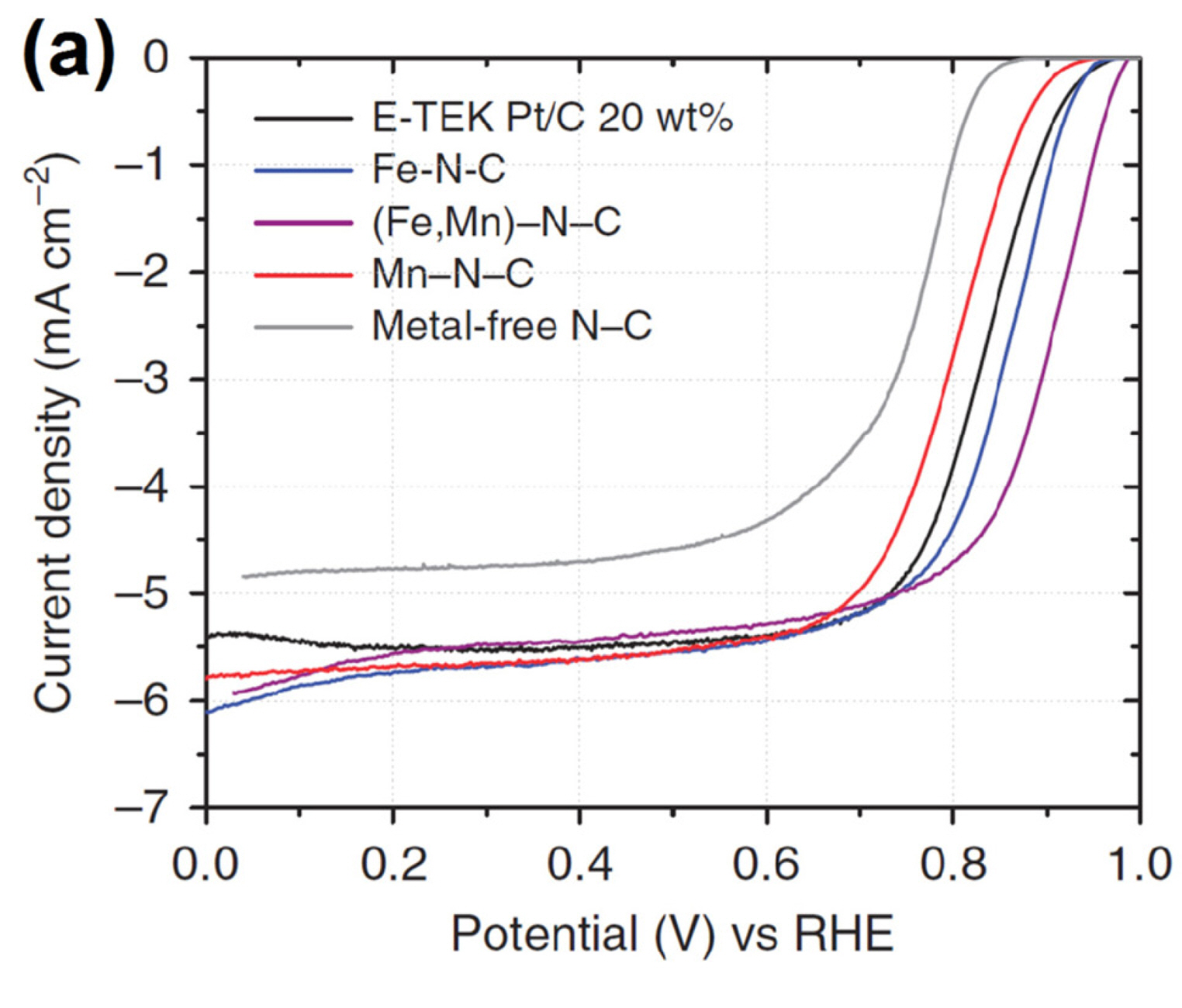
The simplest approach involves the pyrolysis of a mixture containing two transition metal precursors. Some examples have demonstrated that bimetallic FeM−N/C catalysts outperform monometallic Fe−N/C catalyst [26,30,62,77,78]. Zelenay and co-workers reported a highly active FeCo−N/C catalyst produced by the pyrolysis of Fe and Co ions and PANI complex. The prepared FeCo−N/C catalyst exhibited better activity than Fe−N/C and Co−N/C catalysts [26]. Similar synergistic effect was demonstrated by Strasser et al. with FeMn−N/C catalysts, which were prepared from a mixture of Fe- and Mn-PANI (Fig. 7(a)). Mössbauer spectroscopy suggested the absence of alloy-like interactions between Fe and Mn sites in their FeMn−N/C catalyst. However, temperature-programmed desorption of CO revealed changes in the shift of the desorption temperature peak, which indicates that the introduction of the secondary metal during the preparation of Fe−N/C catalysts might modify the geometric and electronic structures of active sites [62].
Joo and co-workers developed a bimetallic FeCo−N/C catalyst with abundant Fe−Nx and Co−Nx sites, organized in ordered mesoporous porphyrinic carbon frameworks (FeCo-OMPC). The bimetallic catalyst showed synergistically enhanced ORR activity compared to those of monometallic catalysts, and its activity compared favorably to the activity of Pt/C in acidic media (Fig. 7(b)) [30]. This bimetallic synergy was corroborated by DFT calculations of O binding energy that was optimized in the presence of bimetallic sites. In addition, EXAFS analyses together with DFT calculations suggested a plausible structure of the bifacial active Fe−Nx sites (inset of Fig. 7(b)). Adsorption of O2 molecules in the interlayer creates a unique bridge-like intermediate structure (Fe−O−O−Fe), which may facilitate effective O−O bond breakage. Interestingly, EXAFS and high-resolution TEM results consistently revealed that the interlayer spacing within the porphyrinic carbon framework was around 4.8 Å, which is similar to the Fe−Cu distance in CcO as well as the Co−Co distance in Collman’s active bifacial porphyrin molecular catalyst [79]. The similarities between the local environment of FeCo-OMPC and those of enzymatic and molecular catalysts may exert a significant role in boosting its ORR activity.
Fig. 7.
(a) ORR polarization curves for (Fe,Mn)-PANI catalysts in 0.1 M KOH. Reprinted with permission from Ref. [62].
(b) ORR Tafel plot for (Fe,Co)-OMPC catalysts measured in 0.1 M HClO4. Inset of (b) represents the bifacial local structures of Fe−Nx and Co−Nx in FeCo-OMPC. Cyan, yellow, blue, gray, red, and white spheres indicate iron, cobalt, nitrogen, carbon, oxygen, and hydrogen atoms, respectively. Reproduced with permission from Ref. [30].

6. Iron and/or Iron Carbide Particles Encapsulated with Graphitic Carbon Shells (Fe−Fe3C@C)
Due to the high-temperature pyrolysis step performed during the preparation of Fe−N/C catalysts, metallic iron and iron carbide (Fe3C) phases stable at high temperature are generated unavoidably along with Fe−Nx sites. At a sufficiently high temperature and in the presence of C and H sources, these metallic Fe−Fe3C particles catalyze the Fischer-Tropsch process in situ, thereby inducing the deposition of graphitic carbon shells on the metallic Fe−Fe3C surface, and generating core−shell-type Fe−Fe3C@C particles. Recently, several groups raised the possibility that Fe−Fe3C@C species might serve as effective active sites for ORR [31,48,49,80-82]. Moreover, Fe−Fe3C@C-based catalysts exhibited excellent durability owing to the presence of outer carbon layers, which can prevent catalyst degradation induced by the radicals produced via Fe-catalyzed Fenton reactions [49,82].
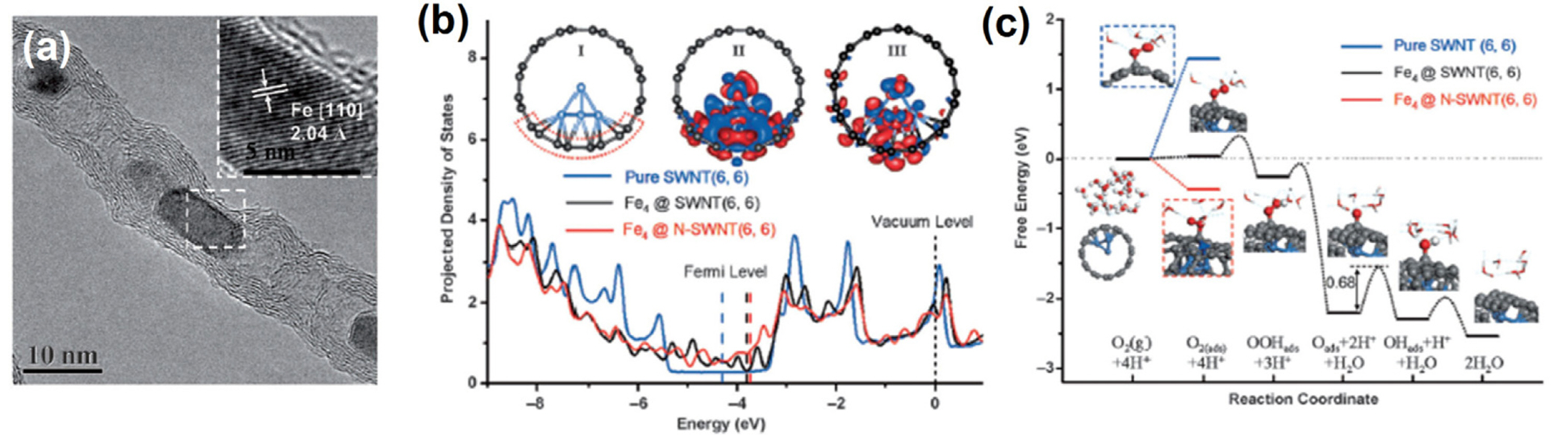
Bao et al. were the first to suggest that Fe−Fe3C@C species might be the active species for the ORR [48]. They synthesized Fe NPs embedded in peapod-like CNTs (Pod-Fe). In acidic media, Pod-Fe exhibited better ORR activity than bare CNTs, and its ORR activity was further enhanced through N-doping. Their encapsulated catalyst was not poisoned by CN− ions, as evidenced by the essentially identical ORR activity before and after the addition of CN−. This result confirmed that the Fe-related species in the catalysts did not directly participate in the ORR. Through DFT calculations and ultraviolet laser enhanced photoemission electron microscopy, they showed that the electron transfer from the inner Fe NPs to CNTs leads to a decreased local work function of the carbon surface, which in turn activates the carbon surface for the ORR. Li and co-workers prepared Fe3C NPs embedded in hollow carbon spheres (Fe3C/C) through a single-step pyrolysis process utilizing ferrocene and cyanamide under high pressure [49]. In the prepared catalyst, the surface of Fe3C NPs coated with graphitic carbon layers was uniformly dispersed with very low quantities of nitrogen or iron functional groups, therefore suggesting that the participation of Fe−Nx moieties in electrocatalysis is negligible. The ORR activity was significant in alkaline media in spite of near complete absence of available Fe−Nx sites on the catalyst surface, which implies that the carbon shell encapsulating Fe3C NPs contributed directly to the high ORR activity. Controlled destruction of Fe3C@C species suggested that the Fe3C@C species act as the active sites for ORR. Gewirth et al. developed a post-treatment method applicable to Fe−N/C catalysts to selectively remove Fe−Nx sites [82]. Their as-prepared Fe−N/C catalyst was composed of both Fe−Nx and Fe−Fe3C@C species. Heat-treatment under Cl2 gas at 900 °C led to almost complete conversion of Fe-based sites into FeClx-like species, accompanied by significant decrease in ORR activity. However, following additional heat-treatment under H2 gas at 900 °C, the ORR activity was fully recovered. XAS and Mössbauer spectroscopy revealed the H2-treated catalyst consisted exclusively of Fe NPs encapsulated with graphitic carbon. Taking these results into consideration, Fe@C was suggested as the active species in the ORR.
Some groups have proposed the existence of a synergetic effect between Fe−Nx and Fe−Fe3C@C sites for the ORR [83-86]. Wan and co-workers prepared three types of catalysts with different active species by controlling the content of Fe in the precursor. The catalyst with the lowest Fe content (Fe@C-FeNC-1) consisted exclusively of Fe−Nx sites [86]. A higher content of Fe in the precursor mixture resulted in catalysts (Fe@C-FeNC-2 and Fe@C-FeNC-3) with both Fe−Nx sites and metallic Fe−Fe3C species. The XPS spectra revealed that the peak associated with Fe−Nx sites in Fe@C-FeNC-1 shifted by 0.7 eV to a higher binding energy than that in Fe@C-FeNC-2 and Fe@C-FeNC-3, suggesting that interactions exist between Fe−Nx and Fe−Fe3C@C species. Fe@CFeNC-2 and Fe@C-FeNC-3 showed ORR activities superior to that of Fe@C-FeNC-1. These results indicated that Fe−Fe3C NPs may synergistically boost the activity of Fe−Nx sites.
There is an on-going debate on the role of Fe−Fe3C@C sites in the ORR, and some recent studies have tried to resolve this issue. Mukerjee et al. prepared an Fe−N/C catalyst and treated it with peroxide, which decreased the number of Fe-based particles [55]. The reduced number of Fe−Fe3C@C sites in the Fe−N/C catalyst resulted in a significant decline in ORR activity in acidic media, but only a negligible effect in alkaline media. In addition, CN− poisoning did not affect the number of electrons transferred during the ORR in an acidic electrolyte, but resulted in a dramatic increase in the production of OH2− in an alkaline electrolyte. They suggested that Fe−Fe3C@C sites play an important role in reducing the H2O2 produced from Fe−Nx sites under acidic conditions, and only a minor role under basic conditions. Recently, Joo and co-workers investigated the role of Fe−Fe3C@C sites in Fe−N/C catalysts using model catalysts [87]. In order to minimize the heterogeneity of the active species, they exploited the phase conversion method starting from Fe3O4 NPs supported on CNTs. The resulting model catalysts contained selectively Fe−Nx, Fe−Fe3C@C, or N-doped carbon (C−Nx) sites. In this work, CN− poisoning and an electrochemical HO2− reduction experiment revealed that the Fe−Fe3C@C sites catalyze predominantly the 2-electron reduction of HO2−, whereas Fe−Nx sites play a major role in catalyzing the 4-electron reduction of oxygen.
Fig. 8.
(a) TEM image of Pod-Fe catalyst. (b) Projected density of states and (c) free energy diagram for the ORR of Fe4@SWNT, Fe4@N-SWNT, and pure SWNT obtained by DFT calculations. Reprinted with permission from Ref. [48].
(d) TEM image of Fe3C/C-700 catalyst. (e) XRD patterns of Fe3C/C-700 before and after single step acid-leaching treatment (tagged with L) and double step ball milling and acid leaching (tagged with BL). (f) ORR polarization curves of Fe3C/C-700 catalyst measured in 0.1 M HClO4. Reprinted with permission from Ref. [49].

7. Conclusions and Outlook
This topical review addressed the recent advances in the understanding of active sites in pyrolyzed Fe−N/C catalysts and their correlations with ORR activity. A growing body of work based on advanced spectroscopic and electrochemical methods suggests that Fe−Nx sites, more specifically, distorted Fe−Nx sites, play a major role in catalyzing ORR. Accordingly, new synthetic strategies have been developed that can produce Fe−Nx sites, preferentially or exclusively, which can lead to the enhancement in the ORR activity. Further efforts have been directed at preparing bimetallic FeM−N/C catalysts that might be able to mimic Nature’s enzyme, CcO. In this manner, researchers are exploiting bimetallic synergism to boost the ORR activity. The catalytic role of the recently reported new type of active sites, namely Fe−Fe3C@C sites, was reviewed. Although some groups suggested the promoting effect of Fe−Fe3C@C species on ORR, the debate over the catalytic role of these species remains on-going.
On the basis of the above-discussed progress achieved in the understanding of catalytic active sites in pyrolyzed Fe−N/C catalysts, a number of high-performance ORR catalysts have been prepared. Some Fe−N/C catalysts showed highly promising ORR activities in acidic media, which approach that of the Pt/C catalyst. In alkaline media, notable Fe−N/C catalysts were shown to even outperform Pt/C. However, we note that such outstanding ORR activities and durability performances are generally, with only few exceptions, obtained with half-cell configurations using rotating disk electrode (RDE) measurements. Hence, significant future efforts are necessary to fully translate the highly promising ORR activities in the RDE measurements into single-cell performances in the membrane electrode assembly.




