


1. Introduction
Charge transfer characteristics of organic semiconductors are essential for the understanding and design of organic light-emitting devices (OLEDs) that are generally composed of various stacked organic film layers, with energetic characteristics and interlayer charge transfer kinetics being highly important for understanding their performance [1-3]. Voltammetry is a common method for determining the energetic characteristics of molecules contained in the above films [4]. In particular, cyclic voltammetry (CV) provides information on the formal oxidation potential (Eo’), which is indirectly correlated to the energy level of the highest occupied molecular orbital (HOMO). However, the seemingly easy determination of energetic parameters often leads to misinterpreted voltammetric data and scientific misconduct [5]. Voltammetric methods can be used to estimate various electron-transfer parameters formal oxidation potential (Eo’), HOMO energy level (EHOMO), electron transfer rate constant (ko’), and oxidation diffusion coefficient (Do), but careful consideration in a limited condition is required [6].
In this study, voltammetric analyses were utilized to characterize electron transfer in iridium(III) bis(2-phenylpyridinato-N,C2’)acetylacetonate ((ppy)2Ir(acac)), a representative green dopant for organic light-emitting devices (OLEDs) (Fig. 1), affording parameters such as Eo’, EHOMO, ko’, and Do. Due to the quasi-reversible electrochemical oxidation of (ppy)2Ir(acac), the above thermodynamic and even kinetic properties could be easily estimated. CV, chronocoulometry (CC), and the Nicholson method were used, and an in-depth explanation of voltammetric techniques is provided.
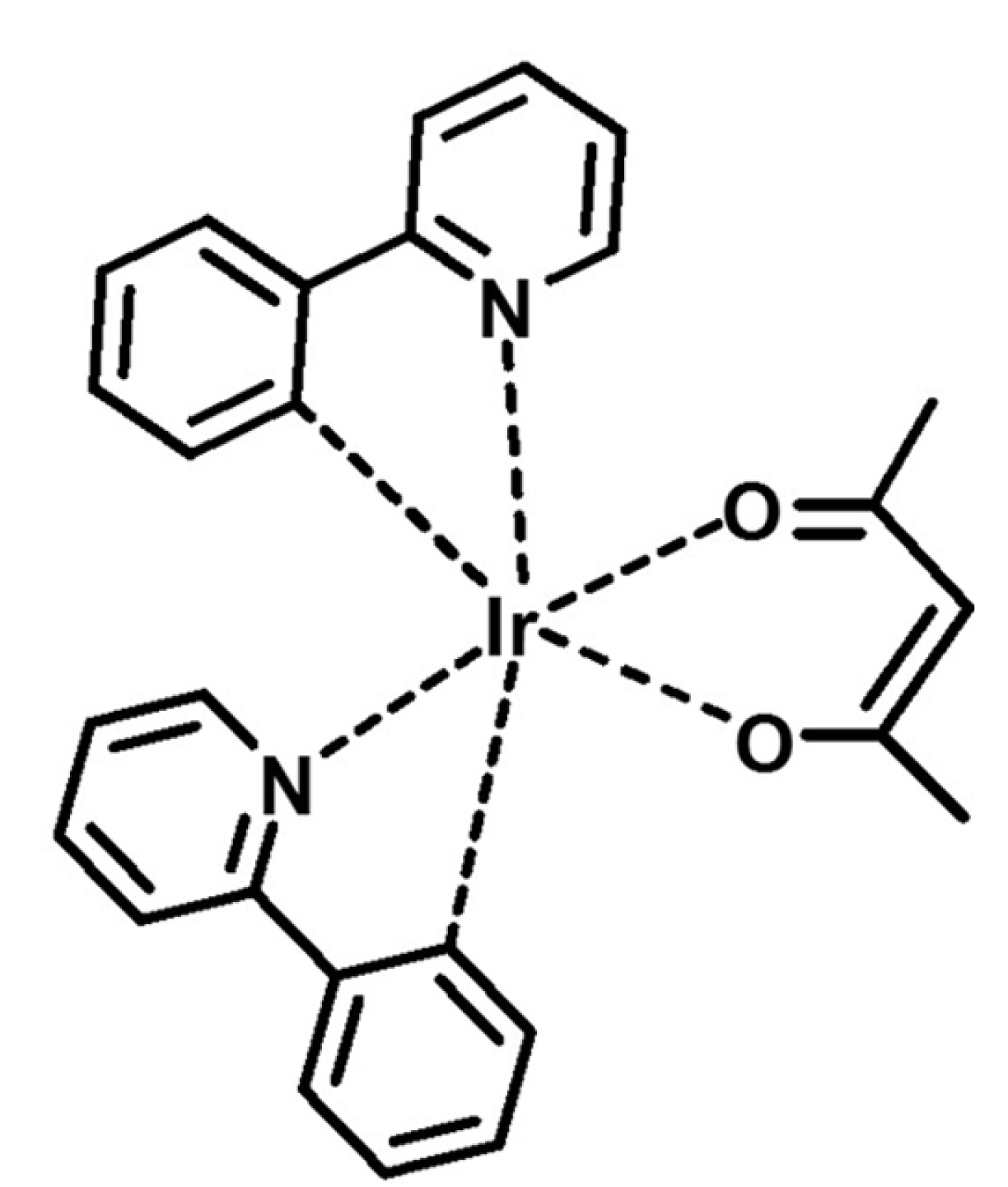
2. Experimental Section
1H and 13C NMR spectra were recorded using an AV-300 (Bruker, Germany) NMR spectrometer. Although the investigated Ir(III) complexes were air-stable, all related manipulations were carried out under nitrogen due to the possible oxidation and thermal decomposition of transient intermediates. Electrochemical characterization was performed using a CH Instruments 650B analyzer (CH Instruments, Inc., TX, USA). Individual solutions were characterized using CV and CC to determine the electron transfer characteristics of (ppy)2Ir(acac).
2.1 Chemicals
All reagents were purchased from either Sigma–Aldrich (Sigma-Aldrich Corp., St. Louis, MO, USA) or TCI (Tokyo Chemical Industry, Tokyo, Japan), except for IrCl3·3H2O (STREM Chemical Inc., MA, USA). All chemicals were used without further purification.
2.2 Synthesis of [(ppy)2Ir(μ-Cl)]2
The cyclometalated Ir(III) μ-chloro-bridged dimer, (ppy)2Ir(μ-Cl)2Ir(ppy)2, was synthesized by the method reported by Nonoyama [7], which involved refluxing IrCl3·3H2O with two equivalents of the cyclometalating ligand in a 3:1 mixture of 2-ethoxyethanol and water. The target product was obtained as a yellow precipitate upon cooling the reaction mixture to room temperature.
2.3 Synthesis of (ppy)2Ir(acac)
The synthesis of (ppy)2Ir(acac) was performed according to a previous report [8]. Briefly, [(ppy)2Ir(μ-Cl)]2 (0.078 mmol), acetylacetone (0.2 mmol), and Na2CO3 (90 mg) were refluxed in 2-ethoxyethanol for 12-15 h in an inert gas atmosphere. After cooling the mixture to room temperature, the produced colored precipitate was filtered off and washed with water and two portions of hexane and ether. The crude product was purified by flash column silica gel chromatography (CH2Cl2) to afford (ppy)2Ir(acac) in 80–90% yield after evaporation and drying. 1H-NMR (CD2Cl2, 300 MHz): δ 8.53 (d, J = 5.7 Hz, 2H), 7.91 (d, J = 8.0 Hz, 2H), 7.81 (t, J = 7.3 Hz, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.22 (t, J = 6.1 Hz, 2H), 6.88 (t, J = 7.3 Hz, 2H), 6.72 (t, J = 7.3 Hz, 2H), 6.27 (d, J = 7.6 Hz, 2H), 5.34 (s, 1H), 1.84 (s, 6H).
2.4 Electrochemical characterization
The electrochemical redox behavior of individual solutions was investigated using CV and CC. The studied solutions commonly contained 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAPF6) in HPLC-grade acetonitrile as a supporting electrolyte and were purged with ultrapure N2 before measurements. A 3-mm-diameter glassy carbon (GC) electrode was employed for CV experiments, and a Ag/Ag+ reference electrode (3 M AgNO3) was used. The GC working electrode was polished with 0.05-μm alumina (Buehler, IL, USA) on a felt pad, followed by 5-min sonication in a 1:1 mixture of deionized water and absolute ethanol. The sonicated electrode was blown dry with N2 gas for 1 min.
3. Results and Discussion
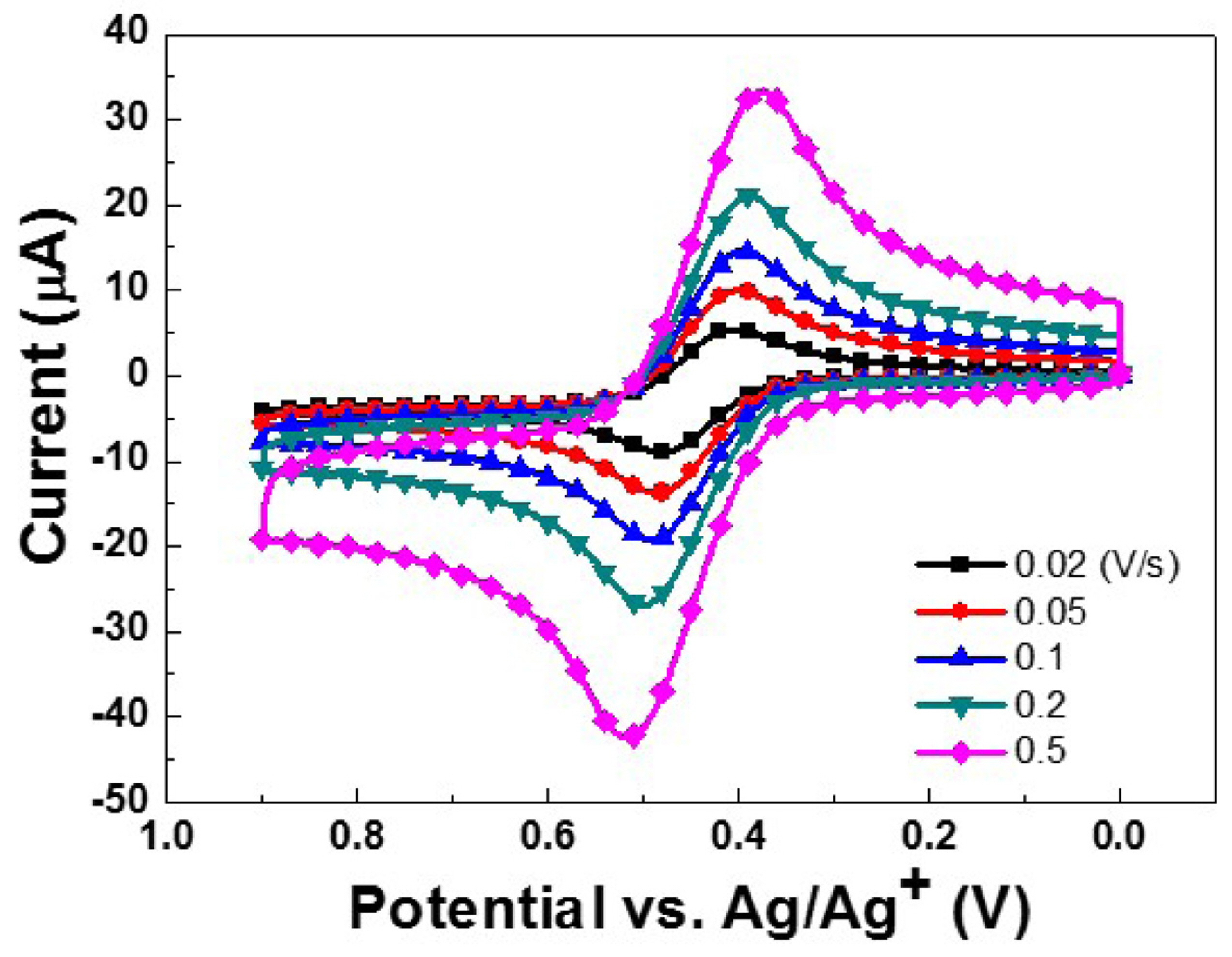
Prior to the characterization of (ppy)2Ir(acac), CV measurements were performed for the ferrocene/ferrocenium (Fc0/+) couple (1.0 mM) in acetonitrile. The formal oxidation potential of Fc was determined as Eo’(Fc0/+) = 0.090 V vs. Ag/Ag+ and was later used as an internal reference for estimating the EHOMO of (ppy)2Ir(acac). CV measurements were subsequently performed for a 1.0 mM solution of (ppy)2Ir(acac), which exhibited quasi-reversible oxidation (Fig. 2) corresponding to the redox conversions of the (ppy)2Ir(acac)0/+ couple [9]. The formal oxidation potential of the above couple was determined as Eo’((ppy)2Ir(acac)0/+) = 0.444 V (vs. Ag/Ag+), being almost equal to the value reported previously [10]. At a scan rate (ν) of 20 mV/s, the (ppy)2Ir(acac)0/+ couple showed a peak potential separation (ΔEpp = Epa − Epc) of 77 mV, indicating a stoichiometric number of transferred electrons (namely one) in each process. With increasing ν (from 20 to 500 mV/s), ΔEpp gradually increased, whereas Eo’((ppy)2Ir(acac)0/+) remained constant, and the ratio of anodic and cathodic peak currents (ipc/ipa) slightly decreased from unity.
Fig. 2.
(a) Cyclic voltammograms of 1.0 mM (ppy)2Ir(acac) recorded at various scan rates (ν) (0.10 M TBAPF6, GC working electrode, Pt counter electrode, and Ag/Ag+ reference electrode in acetonitrile solution).
According to Forrest et al. [11], the formal potential of (ppy)2Ir(acac)0/+ is related to its EHOMO:

where ECV is the relative oxidation potential of (ppy)2Ir(acac)0/+ calibrated against the Fc0/+ couple. The ECV of (ppy)2Ir(acac)0/+ was calculated as 0.354 V (= 0.444 − 0.090), and EHOMO was estimated as −5.10 eV, being identical to previously reported values determined by ultraviolet photoemission spectroscopic (UPS) measurements [12].
The oxidative diffusion coefficient (Do) of (ppy)2Ir(acac) was also determined based on CC measurements. In our approach, a double-step potential was applied to the solution, inducing an instantaneous oxidation of (ppy)2Ir(acac)in the vicinity of the electrode in the forward step, followed by rapid reduction of the oxidized form to (ppy)2Ir(acac) in the reverse step. Potentials sufficiently more positive than 0.444 V were applied in the forward step (and vice versa), and almost identical charge (Q) vs. time (t) profiles were obtained at |Eapp − Eo’| = 0.591 V (Fig. 3a). The applied overpotential accompanied the changes of the ratio of the concentration of (ppy)2Ir(acac) and (ppy)2Ir(acac)+ at the surface of electrode. According to the Nernst equation, the [(ppy)2Ir(acac)]/[(ppy)2Ir(acac)+] ratio at the electrode surface rapidly changed to 1/1010 under the condition of Eapp − Eo’ = 0.591 V, with the diffusion of (ppy)2Ir(acac) being the only mode of subsequent mass transport. A strictly linear relationship between Q and t1/2 (y = 1.34 × 10−5·x, R2 = 0.999) was obtained (Fig. 3b), and Do was calculated using the Anson equation:
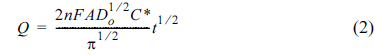
where n is the number of electrons, A is the electrode area, and C* is the bulk concentration of (ppy)2 Ir(acac).
Fig. 3.
(a) CC curve of 1.0 mM (ppy)2Ir(acac) and (b) typical Anson correlation obtained from (a). Two-step potentials were applied, inducing instantaneous oxidation of (ppy)2Ir(acac) in the first step followed by reversal in the second step (|Eapp−Eo’| = 0.591 V).
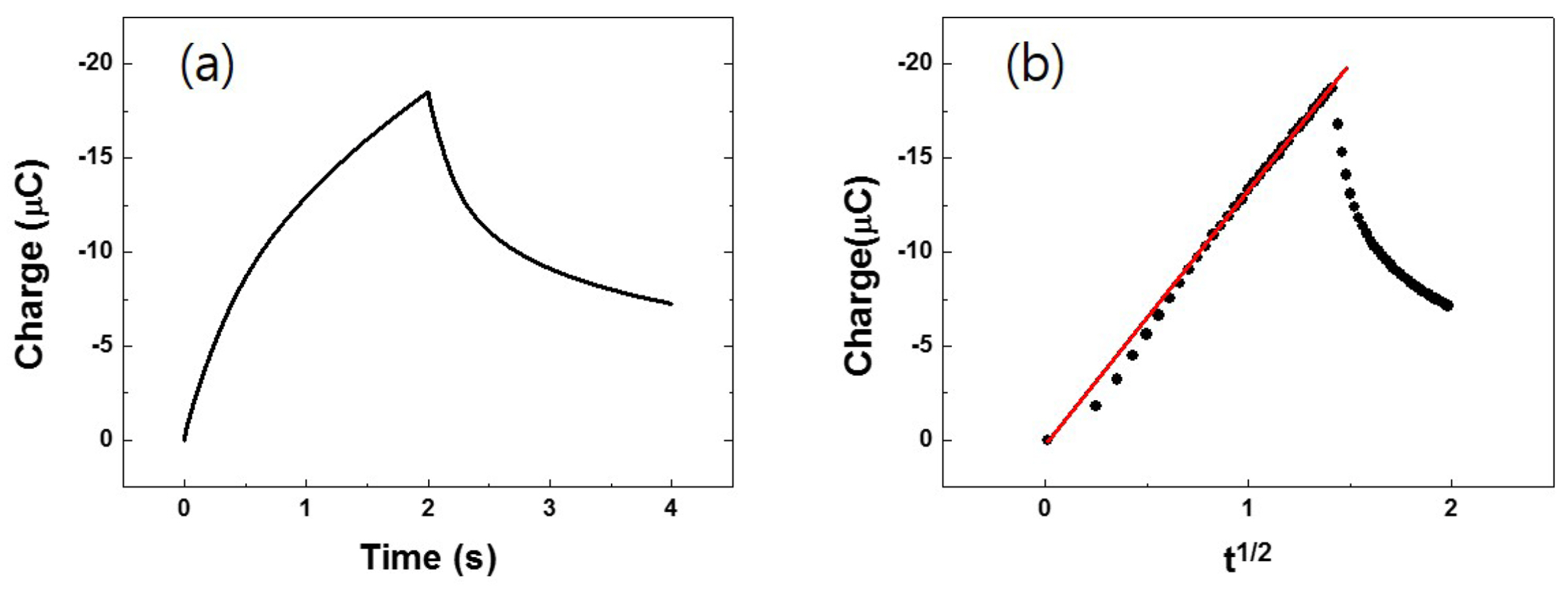
The A value of glassy carbon was previously measured as 7.64 × 10−2 cm2 using 1.0 mM Fc in acetonitrile (not shown here), and thus, the diffusion coefficient of (ppy)2Ir(acac) was determined as Do = 1.35 × 10−5 cm2 /s, being slightly lower than that of Fc under similar conditions (2.4 × 10−5 cm2/s). CV data pertaining to reversible and quasi-reversible electron transfer not only provides thermodynamic information, but also allows the estimation of kinetic parameters for the redox process. For diagnostic purposes, Nicholson et al. [13] suggested using a dimensionless kinetic parameter Ψ, which is a function of ν, Do, and ko’:
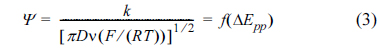
The quasi-reversible one-electron oxidation exhibited by (ppy)2Ir(acac) implies that ΔEpp is close to 59 mV for slow potential scan rates, increasing concomitantly with the scan rate in the case of fast-scan CV measurements. According to Nicholson’s method, Ψ is correlated with ΔEpp (Fig. 4a) [14] and can therefore be determined by measuring ΔEpp values at various scan rates. As shown in Fig. 4b, Ψ values were extracted from CV curves at various scan rates, being linearly dependent on ν−1/2 (y = 1.80 × 10−1·x, R2 = 0.985). Finally, the ko’ of (ppy)2Ir(acac) was calculated as 2.68 × 10−2 cm/s based on eq. (3), being comparable to that of the Fc0/+ couple (1.91 × 10−2 cm/s) under similar conditions [15].
Fig. 4. (a)
(a) Theoretical relationship between the dimensionless kinetic parameter Ψ and peak potential separation (ΔEpp); (b) linear relationship between Ψ and ν−1/2 for (ppy)2Ir(acac). ΔEpp values experimentally determined for several ν values were used to estimate Ψ, which showed a linear dependence on ν−1/2.

4. Conclusions
Two voltammetric techniques, CV and CC, were used to investigate the redox characteristics of iridium(III) bis(2-phenylpyridinato-N,C2’)acetylacetonate ((ppy)2Ir(acac)), a typical green dopant for OLEDs. Although voltammetry is one of the most versatile analytical techniques for the study of electroactive materials, it requires precision, an appropriate level of understanding, and suitable experimental conditions. The quasi-reversible oxidation of (ppy)2Ir(acac) allowed to estimate the HOMO energy levels, and the electron-transfer parameters such as diffusion coefficients (Do) and electron transfer rate constants (ko’) were successfully determined using a combination of CV and CC measurements.




