


1. Introduction
Conventional lithium-ion batteries (LIBs) are the primary energy source for applications in various energy storage systems such as those for portable devices, medical equipment, and transportation systems, including electric vehicles and hybrid electric vehicles, owing to their high energy and power capability, and long cycle life [1-3]. However, the expansion of their uses is hindered by their limited applications and electrochemical performance, and the high cost of materials such as nonaqueous electrolytes, which also have problems such as low safety and the presence of flammable or toxic components. These limitations motivate the further development of next-generation battery systems such as aqueous battery systems, lithium metal batteries, and all-solidstate batteries [4-6]. Among the battery systems currently being considered, aqueous rechargeable lithium-ion batteries (ARLBs), which were first announced by Li et al. in 1994, have the potential to overcome the crucial issues related to cost and safety [7,8]. Furthermore, among all the candidates for stable electrolytes for use in ARLBs, including polymers, ionic liquids, and ŌĆ£water-in-saltŌĆØ electrolytes, aqueous electrolyte solutions in particular have attracted attention owing to their high safety, use of abundant elements, environmental friendliness, and use of inexpensive materials [9-11]. Therefore, the development of high-performance ARLBs may solve these issues and make it possible to replace conventional LIBs in large-scale applications.
In the development of ARLBs with high energy density, the current conventional cathode and anode materials used in ARLBs can provide high electrochemical performance. Widely investigated cathode materials include LiCoO2 [12], LiFePO4 [13], and LiMn2O4 [14]; however, the anode materials for ARLBs are limited owing to the poor electrochemical stability window of aqueous electrolyte solutions compared to in the conventional LIBs. The anodic and cathodic stabilities are only 1.23 and 0.0 V versus standard hydrogen electrode (SHE), respectively, which limits electrochemical performance [7,15]. Furthermore, it should be noted that the poor cycling performance of the anode materials also can be related to the dissolution of the metal-ions, formation of unwanted side reaction components on the surface of the active materials, and irreversible structural transformation and impact of the components of the decomposition of the water that lead to formation of the side reactions components [7,13,16]. However, among alternative anode materials for ARLBs, LiV3O8 has garnered attention because of its high theoretical capacity (279 mAh g-1), available operating potential, low cost, and safety [17,18]. Although LiV3O8 has been widely investigated, its cycling performance is limited owing to low electronic conductivity, irreversible structure phases, dissolution of various vanadium ions, and formation of unfavorable side reactions components. These characteristics lead to capacity fading and limit its electrochemical performance in aqueous electrolyte solutions owing to the narrow electrochemical stability window of water, hindering application of LiV3O8 in LIBs and ARLBs [19,20]. Therefore, to overcome these issues, many researchers have applied various approaches to enhance the electrochemical performance of LiV3O8 by doping [20] and application of surface coatings, such as carbon [21], polymers [22], solid and ceramic materials [23,24], and Al2O3 [25]. For ARLBs, in particular, we suggest that surface coatings can play an important role in stabilizing the structure of LiV3O8, reducing dissolution of metal ions, and preventing the formation of side reaction components on the surface of the active materials. In addition, as shown in previous works [13,14,26-32], the metal fluorides AlF3, LaF3, and MgF2 have strong ionic bonding with high ionic conductivity and are also chemically and electrochemically stable, resulting in significant application of these materials as surface agents for surface stabilization of cathode and anode materials so that lithium-ion transport during cycling is not prevented. Thus, herein we report application of an AlF3 coating to improve the surface stability and cycling stability of the anode material LiV3O8 in aqueous electrolyte solutions.
2. Experimental
To synthesize LiV3O8, Li2CO3 (Aldrich) and V2O5 (Aldrich) were mixed in a 1:3 molar ratio and subjected to a solid-state reaction at 680Ōäā for 24 h in air. A chemical deposition method was used to obtain AlF3-coated LiV3O8 samples. The as-prepared LiV3O8 powder (ŌĆ£bare LVOŌĆØ hereafter) was immersed in an Al(NO3)3 9H2O aqueous solution, and an NH4F aqueous solution was added. The molar ratio of Al to F was controlled at 1:3, and the designed amount of AlF3 was 1, 2, or 3 wt.% of the amount of LiV3O8 electrode material; the resulting samples are denoted as AlF3-1-LVO, AlF3-2-LVO, and AlF3-3-LVO, respectively. The mixed solutions were dried at 120Ōäā for 1 h in an oven. The asobtained samples were calcined at 400Ōäā for 2 h in an argon flow.
The crystalline structures of the synthesized LiV3O8 and AlF3-coated LiV3O8 materials were determined by X-ray diffraction (XRD) analysis using a Rigaku SmartLab diffractometer with Cu K╬▒ radiation (40 kV, 250 mA) in a 2╬Ė range of 10-80┬░ at a scan rate of 0.03┬░ s-1. The surface morphologies of these samples were observed using field emission scanning electron microscopy (FE-SEM, JSM 7800F) with energy-dispersive X-ray spectroscopy (EDS). To obtain the surface chemical states of the as-prepared and cycled samples, X-ray photoelectron spectroscopy (XPS) analysis was performed using a Physical Electronics PHI5000 VersaProbe II instrument with an X-ray Al anode as the source. To determine the chemical composition of the 1 M Li2SO4 aqueous electrolyte solutions after 100 cycles, the Li and V concentrations of bare and AlF3-coated LVO samples were measured by inductively coupled plasma (ICP) optical emission spectrometry (Thermo Scientific iCAP 7000, series ICP-OES).
The working electrode (WE) was prepared using as-obtained materials, acetylene black and poly(tetrafluoroethylene) (Aldrich) at a weight ratio of 80:10:10. The black slurry was uniformly mixed by hand for about 20 min and then coated on a stainless mesh of 0.25 cm2 at a loading of 2 mg cm-2. The counter electrode (CE) of activated carbon (RP-20, Kuraway Chemical) was prepared using the method for the WE described above. A silver chloride electrode (RE-1B, ALS, Ag/AgCl, +0.197 V vs. SHE) was used as the reference electrode (RE). A three-electrode electrochemical cell consisting of the WE, CE, and RE in 1 M Li2SO4 aqueous electrolyte solution with a pH of 7 was used for the electrochemical measurements. The galvanostatic chargeŌĆōdischarge performance of the samples was determined using a WonATech potentiostat/galvanostat instrument at different current rates (1 C = 279 mAh g-1) in the voltage range of ŌłÆ0.6ŌłÆ0.3 V (vs. Ag/AgCl) at room temperature. Cyclic voltammetry (CV) measurements were conducted using the WonATech potentiostat/galvanostat at a scan rate of 1 mV s-1 in the potential range of ŌłÆ0.7ŌłÆ0.5 V (vs. Ag/AgCl). The surface resistance of the cycled bare and AlF3-coated LVO samples was determined by electrochemical impedance spectroscopy (EIS) using a multichannel electrochemical workstation (ZIVELAB, MP1, WonATech) in a frequency range of 100 kHz to 10 mHz at a voltage amplitude of 5 mV.
3. Results and Discussion
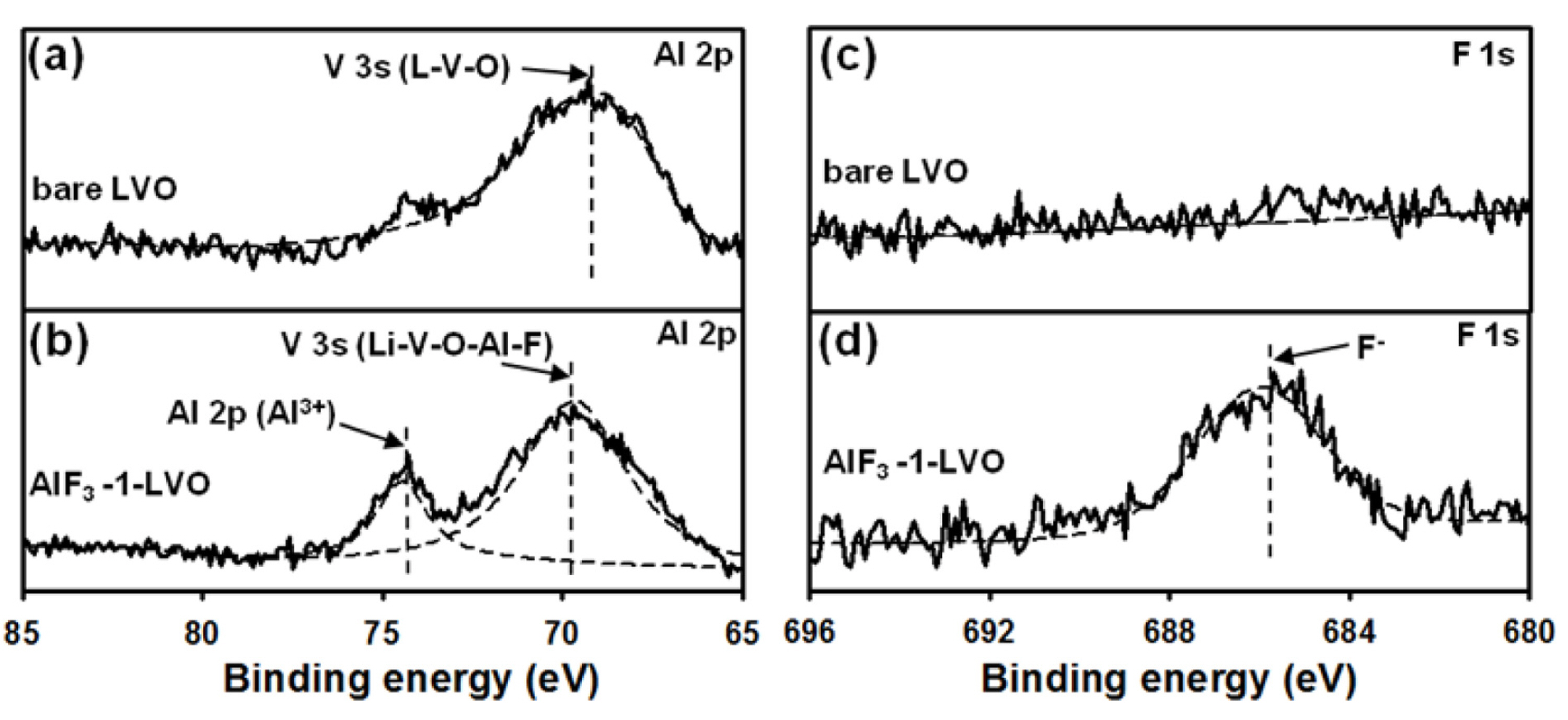
The crystalline structures of the synthesized LiV3O8 and AlF3-coated LiV3O8 materials were identified by XRD, as shown in Fig. 1. The bare LVO sample exhibits XRD patterns showing a monoclinic crystalline structure (JCPDS 72-1193, space group: P21/m). The peak positions of the XRD patterns of the AlF3-coated LiV3O8 materials do not differ from those of bare LVO, and the coating material AlF3 is not observed in the XRD patterns because only a small amount of AlF3 is present on the surface of the LiV3O8 sample. This result shows that coating with AlF3 does not change the structure and the powder phology of the synthesized LiV3O8. Fig. 2 clearly shows that the AlF3-coated LiV3O8 samples have the same surface particles as the bare LVO sample. In addition, to confirm the presence of the AlF3 coating on the surface of the LiV3O8 sample, EDS analysis was conducted (Fig. 3). The EDS spectra indicated the presence of Al and F, confirming the presence of the AlF3 coating; further, the concentrations of these materials increased with increasing coating amount. In addition, the Al 2p and F 1s XPS spectra of these samples are shown in Fig. 4. The Al 2p spectrum of the AlF3-coated LiV3O8 sample consists of two peaks; the peak at 74.41 eV is assigned to Al atoms, and the V 3s peak, at 68.67 eV, may correspond to the formation of a solid solution of Li-V-Al-O-F at the interface between the bare and coated materials [33]. The F 1s peak appears at 685.99 eV; this peak and the Al peak are assigned to the AlF3 coating material. For the bare LVO, the Al 2p and F 1s XPS spectra are not observed; however, the V 3s peak appeared at 68.97 eV and indicates the formation of a solid solution of Li-V-O. Thus, the XPS data are the best to explain the homogeneous coverage by the AlF3 coating on the LiV3O8. Thus, the AlF3 coating was successfully prepared.
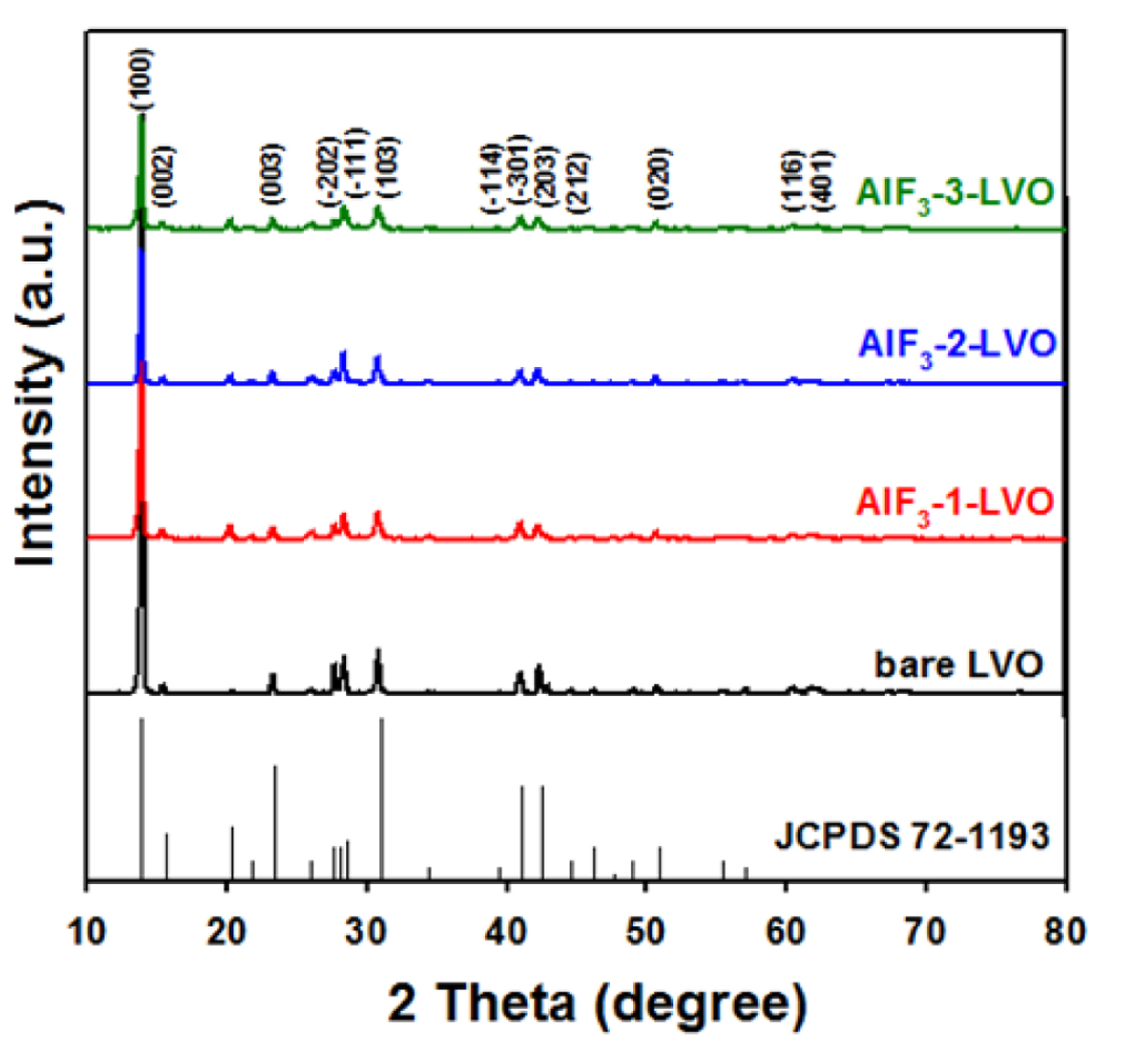
Fig.┬Ā2.
SEM images of (a) bare LiV3O8 (LVO), (b) AlF3-1-LVO, (c) AlF3-2-LVO and (d) AlF3-3-LVO electrodes.

Fig.┬Ā3.
EDS analysis of (a) bare LiV3O8 (LVO), (b) AlF3-1-LVO, (c) AlF3-2-LVO and (d) AlF3-3-LVO electrodes.

Fig.┬Ā4.
(a, b) Al 2p and V 3s and (c, d) F 1s XPS spectra of bare LiV3O8 (LVO) and AlF3-1-LVO electrodes before cycling.
To understand the effect of the AlF3 coating on insertion and extraction of lithium ions into and from the LiV3O8 structure, CV experiments were conducted at a scan rate of 1 mV s-1 in the potential range of ŌłÆ0.7ŌłÆ0.5 V (vs. Ag/AgCl). The CV curves for the bare LVO and AlF3-1-LVO electrodes are presented in Fig. 5a. The CV curves of the samples have similar shapes and two peaks that correspond to the intercalation and deintercalation processes. It should be noted that CV curves have one current peak located at approximately ŌłÆ0.6 V, which corresponds to the intercalation process and two current peaks at approximately ŌłÆ0.2 and 0.25 V (vs. Ag/AgCl) for the deintercalation of the lithium-ions, which is in good agreement with the cyclic performance of LiV3O8 electrodes as shown in Fig. 5. The intercalation and deintercalation process of the lithium ions into/from the structure of the LiV3O8 material in the nonaqueous and aqueous electrolytes is accompanied by the phase transformation of the LiV3O8 (Li1+xV3O8), which occurs during the electrochemical processes [34,35]. Therefore, the LVO electrode in nonaqueous electrolytes has several oxidation and reduction peaks that do not appear for the LVO electrode in Fig. 5a, owing to the narrow potential range considering the poor electrochemical stability window of water, which has anodic and cathodic stabilities of 1.23 and 0.0 V versus SHE, respectively [7,12]. Furthermore, the AlF3-1-LVO electrode has a higher peak current density than the bare LVO electrode. This result indicates that the AlF3-coated LVO electrode has a lower energy barrier for diffusion processes, which can enhance the electrochemical performance of LiV3O8.
To fully explain the impact of the AlF3 coating on the surface stability and electrochemical processes of the electrodes, cycling data were obtained at 1 C in the potential range of ŌłÆ0.6ŌłÆ0.3 V in a 1 M Li2SO4 aqueous electrolyte solution (Fig. 5b-d). The bare LVO electrode delivers a high initial discharge capacity of 106 mAh g-1; however, it maintains a poor cycling life, with a capacity of 22 mAh g-1 after 50 cycles. This result indicates that the bare LVO electrode undergoes attack by the aqueous electrolyte solution and dissolution of vanadium ions, which lead to capacity fading. In contrast, the 1, 2, and 3 wt.% AlF3-coated LVO electrodes have initial specific discharge capacities of 104, 90, and 86 mAh g-1, respectively. Moreover, as shown in Figs. 5b and c, the charge-discharge voltage profiles show that the main electrochemical processes are related to the insertion and extraction of lithium ions into and from the LiV3O8 structure, which is consistent with the electrochemical behavior observed in the CV curves(Fig. 5a). Note that the discharge capacity of 60 mAh g-1 for the AlF3-1-LVO electrode is higher than those of the bare LVO, AlF3-2-LVO, and AlF3-3-LVO electrodes, which are 22, 49, and 27 mAh g-1, respectively. It seems that the optimal amount of AlF3 coating material on the surface of the LVO electrode can successfully enhance the surface stability of the electrode and protect it from direct contact with and attack by the aqueous electrolyte solution. We can conclude that the AlF3 coating can prevent vanadium ion dissolution and formation of side reaction components on the surface of the LVO electrode. This indicates that the AlF3-coated LVO electrodes have higher structural stability than bare LVO electrodes. Note that the presence of the AlF3 coating on the surface of the LVO electrode was confirmed by SEM images after repeated charge-discharge cycling for 20 cycles (Fig. 6). Moreover, SEM images of the AlF3-coated LVO electrodes before cycling show almost identical the shape and size to those of pristine LiV3O8 sample (Fig. 2); in addition, the AlF3-coated LVO electrodes have more uniform surfaces than the bare LVO electrode after cycling. Furthermore, the ICP data show a higher concentration of vanadium ions for the bare LVO electrode (1.249 ppm) than for the 1, 2, and 3 wt.% AlF3-coated LVO electrodes (0.613, 0.426, and 0.334 ppm, respectively) in a 1 M Li2SO4 aqueous electrolyte solution (Fig. 5e). Thus, the AlF3 coating process stabilizes the structure of the electrode and reduces the dissolution of vanadium ions, effectively decreasing the contact between the LVO electrode and the aqueous electrolyte solution. Note that the cycling performance of LiV3O8 was improved by an AlF3 coating in a nonaqueous electrolyte for LIBs [16].
Fig.┬Ā5.
(a) CV of bare LiV3O8 (LVO) and AlF3-1-LVO electrodes at a scan rate of 1 mV sŌłÆ1 in the potential range of ŌłÆ0.7ŌłÆ0.5 V, (b) initial (1st) and (c) 20th charge-discharge voltage profiles, (d) cyclic performance of bare LiV3O8 (LVO) and AlF3-coated LVO electrodes at 1 C in the potential range of ŌłÆ0.6ŌłÆ0.3 V in 1 M Li2SO4 aqueous electrolyte solution, (e) ICP analysis of the 1 M Li2SO4 aqueous electrolyte solutions after 100 cycles and V (vanadium ion) concentration of the bare LiV3O8 (LVO) and AlF3-coated LVO materials, (f) Nyquist plots and equivalent circuit of the LVO materials at a selected cycle (10th cycle) in Fig. 3d.


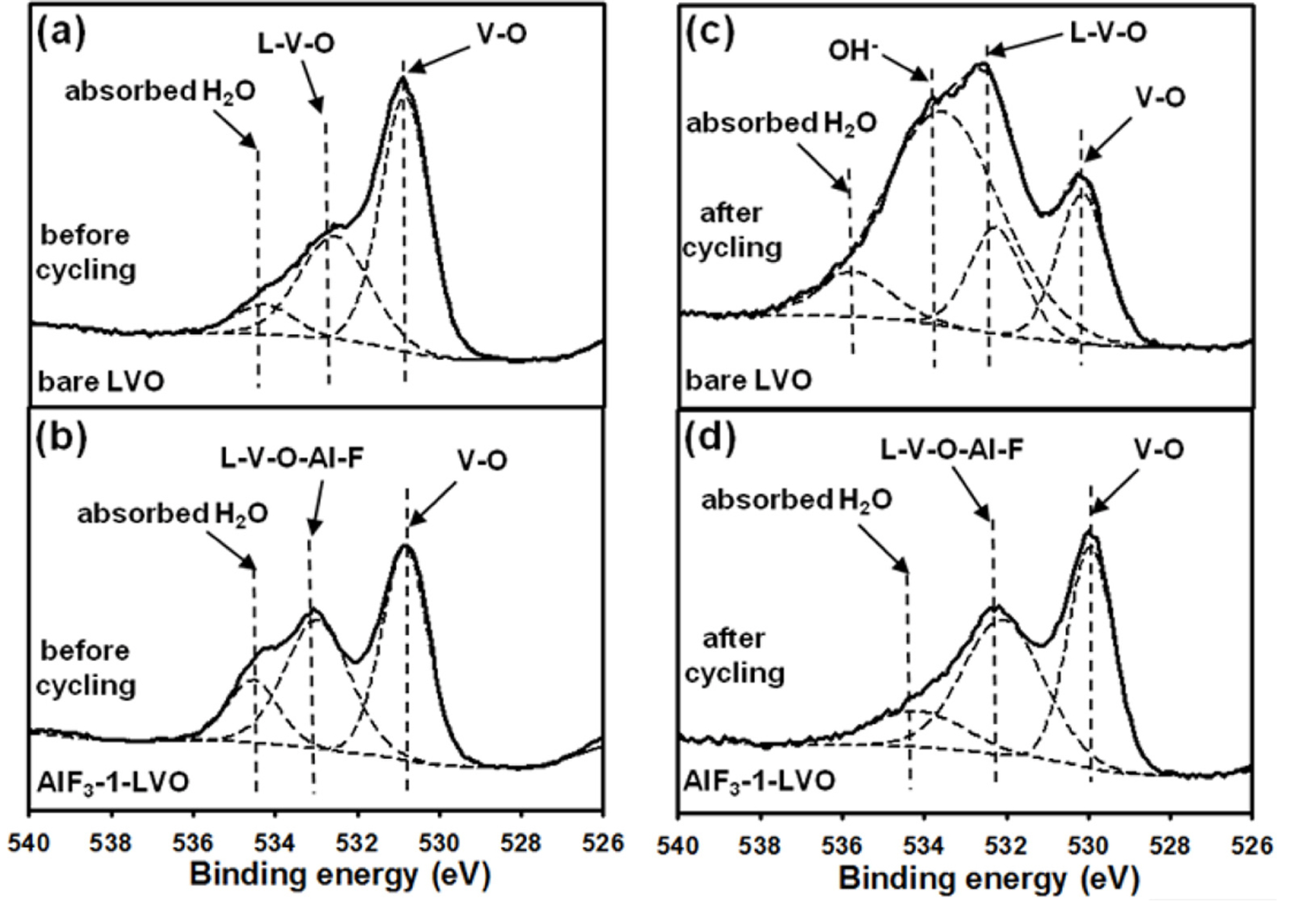
To better understand the reasons for the enhanced electrochemical performance of the AlF3-coated LVO electrodes, the surface resistances of all the samples were obtained using EIS measurements, as shown in Fig. 5f. The Nyquist plots of all the samples were obtained after the 10th cycle and fitted by the equivalent circuit, as shown in Fig. 5f. A more detailed discussion of all the elements affecting the surface stability is presented in our previous work [13,14] The main parameter of the equivalent circuit is the surface resistance (Rsurface); the AlF3-1-LVO sample has a lower value of the resistance (193 ╬®) than bare LVO, AlF3-2-LVO, and AlF3-3-LVO samples (358, 311, and 431 ╬®, respectively). This finding can be attributed to the fact that the AlF3 coating helps reduce surface resistance and improves the diffusion of lithium ions through the interface between the coating and electrode, thus preventing vanadium ion dissolution and formation of unfavorable side reaction components in the aqueous electrolyte solution. Additionally, the surface changes in the bare LVO sample due to decomposition of the side reaction components of the aqueous electrolyte solution following vanadium ion dissolution were investigated by XPS analysis before and after repeated charge-discharge cycling in a 1 M Li2SO4 aqueous electrolyte solution. The O 1s spectra of the bare LVO and AlF3-1-LVO samples before and after cycling in a 1 M Li2SO4 aqueous electrolyte solution are shown in Fig. 7. The corresponding binding energies from the O 1s spectra for bare and AlF3-coated LVO samples are reported in Table 1. Before and after cycling, the XPS spectra of the bare and coated samples have several peaks; for example, the V-O peak is assigned to LiV3O8. The other peaks, L-V-O and L-V-O-Al-F, are assigned to the formation of a solid solution on the interface of the bare material for the bare LVO sample and at the interface between the bare material and the coating for the AlF3-1-LVO sample. Further, a small amount of physically absorbed H2O is present in the samples before and after cycling [36]. Furthermore, the spectrum of the bare LVO sample after cycling has an additional peak corresponding to OHŌłÆ (533.36 eV), which can be attributed to the side reaction components and the surface film on the bare material mentioned above. This peak is not observed for the coated material, which also indicates that the AlF3 coating process significantly affects the surface stability of the LiV3O8 sample.
Fig.┬Ā7.
XPS O 1s spectra of (a, c) bare LiV3O8 (LVO) and (b, d) AlF3-1-LVO samples before and after cycling at 1 C in 1 M Li2SO4 aqueous electrolyte solution, respectively.
Table┬Ā1.
Peak assignment and binding energy from O 1s XPS spectra of bare LiV3O8 (LVO) and AlF3-coated LVO samples before and after cycling.

To further promote the application of bare and AlF3-coated LVO materials, rate capability was examined, as shown in Fig. 8. The AlF3-1-LVO sample maintained a higher specific discharge capacity at various current densities than the other AlF3-coated materials. Further, the specific capacity of the bare LVO sample suddenly decreased from 106 to 1 mAh g-1 as current densities increased from 1 to 20 C. This finding also supports the assertion that the positive effect of the AlF3 coating prevents surface failure and dissolution of vanadium ions, thus leading to a high rate capability.
Fig.┬Ā8.
Rate capabilities of bare LiV3O8 (LVO) and AlF3-coated LVO materials in the potential range of ŌłÆ0.6ŌłÆ0.3 V in 1 M Li2SO4 aqueous electrolyte solution.

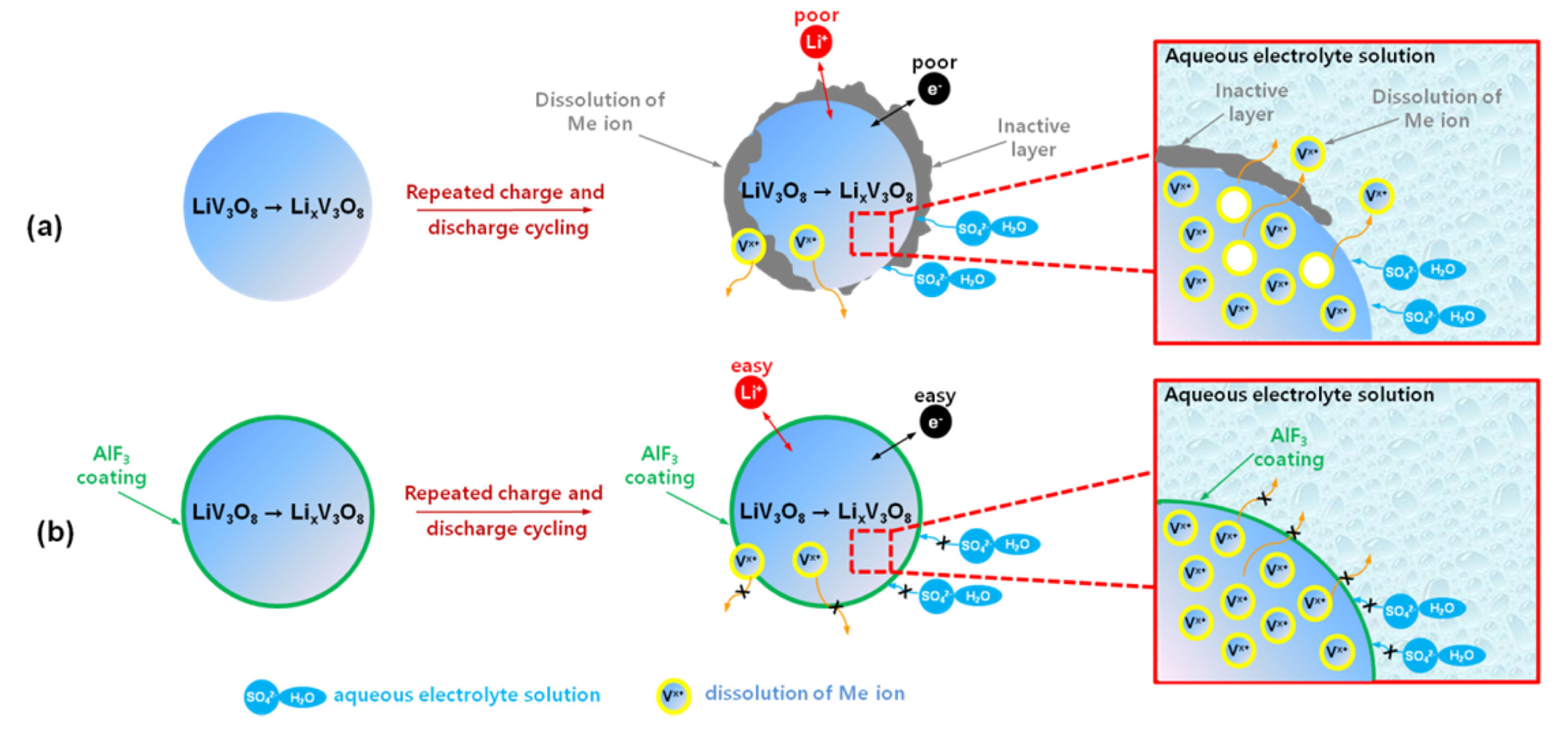
Note that the electrode materials of ARLBs can be soluble in water, which leads to capacity fading [7,13,37]. In particular, materials containing vanadium compounds, such as V2O5, LiV3O8, and LiV2O5, are easily susceptible to dissolution of vanadium ions with various valences in the electrolyte [16,38,39]. Therefore, to overcome this issue, coating technology can be successfully used to improve the surface stability of vanadium-based materials in aqueous electrolyte solutions. In our previous works [13,14], an AlF3 coating was successfully used to overcome the main problem with electrode materials related to the dissolution of metal ions: the formation of unfavorable side reaction components on the surface of the particles and subsequent capacity fading. Therefore, we believe that the AlF3 coating successfully prevented the formation of side reaction components on the surface of the LiV3O8 sample in the aqueous electrolyte solution, improving surface stability, and enhancing electrochemical performance. On the basis of those works [13,14], we propose a schematic model of the failure and improvement of the surface stability of bare LVO and AlF3-coated LVO samples, respectively, in aqueous electrolyte solutions during repeated charge-discharge cycling, as shown in Fig. 9. During cycling, the surface of the bare LiV3O8 sample encounters unwanted side reaction components and attack by the aqueous electrolyte solution, leading to dissolution of vanadium ions and capacity fading during prolonged cycling. Consequently, the polarization and surface resistance increase owing to the formation of an insulating layer that prevents good insertion and extraction of lithium ions into and from the LiV3O8 structure (Fig. 9a). When the AlF3 coating is used as a protective layer, it can contribute to direct, effective stabilization of the surface of the LiV3O8 sample in the aqueous electrolyte solution. In addition, the AlF3 coating on the surface of LiV3O8 particles improves ionic and electronic transfer (Fig. 9b).
4. Conclusions
An AlF3 coating was applied by a simple chemical deposition method as a surface agent for surface stabilization of LiV3O8. The as-coated LiV3O8 material exhibited enhanced cycle life and better rate capability than bare LiV3O8, resulting in reduced vanadium ion dissolution and surface failure, and thus, improved surface stability. The results of various physicochemical and electrochemical investigations confirmed that the electrochemical performance of LiV3O8 was improved by the application of an AlF3 coating with an optimal content of 1 wt.%, which can protect the LiV3O8 from direct attack by the aqueous electrolyte solution, leading to decreased surface resistance and supporting the kinetics of the lithium-ion intercalation and deintercalation processes. Thus, we can suggest that the AlF3 coating has potential application for surface stabilization of LiV3O8 in aqueous electrolyte solutions.




