Recent Progress on Polymeric Binders for Silicon Anodes in Lithium-Ion Batteries
Article information
Abstract
Advanced polymeric binders with unique functions such as improvements in the electronic conduction network, mechanical adhesion, and mechanical durability during cycling have recently gained an increasing amount of attention as a promising means of creating high-performance silicon (Si) anodes in lithium-ion batteries with high energy density levels. In this review, we describe the key challenges of Si anodes, particularly highlighting the recent progress in the area of polymeric binders for Si anodes in cells.
1. Introduction
Although lithium-ion batteries (LIBs) for portable electronic devices have been successfully commercialized, a significant improvement in the energy density of LIBs is required to satisfy the requirements of high power and/or capacity levels for applications such as power tools and electric vehicles and for the efficient use of renewable types of energy [1-5]. To achieve high-energy-density LIBs, silicon (Si) has been extensively investigated as a negative-electrode (anode) material due to its high theoretical specific capacity of 3580 mAh g−1 for Li15Si7 [6-10]. However, the practical application of Si to LIBs is quite challenging because Si is associated with significant volume changes which arise during lithium insertion and extraction, which can hasten mechanical fracture of the electrode, cause a loss of electrical conduction paths, and results in continuous electrolyte decomposition at the active surface when exposed by the cracking of Si during cycling, as depicted in Fig. 1 [11-14].
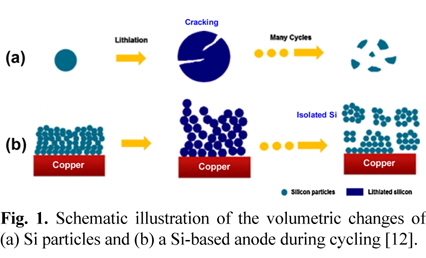
To suppress the volume changes of Si-based anodes, nanostructured anode materials [11,12], nanocomposites of anode materials, porous structures to mitigate the disintegration of Si particles due to colossal volume changes, and a proper electrode design to optimize structural factors such as the particle size, space between the particles, and polymeric binders have been explored to develop anodes that undergo less of a volume change [15,16]. In particular, attention has been devoted to the development of functional polymeric binders, as the polymer binder is a material that is very important for the binding of the Si-active materials of the electrodes. The use of a functional polymeric binder in Si-based anodes is expected to prevent electrical contact loss between the Si particles and to accommodate severe volume changes (ca. 400%) during cycling [17-19]. Recent results have shown that sodium carboxymethyl cellulose (CMC) and poly(acrylic acid) (PAA) binders improve the electrochemical performance of Si-based anodes compared to poly(vinylidene fluoride) (PVDF) binders [20,21]. It is believed that a robust polymeric binder is a promising means of inhibiting the mechanical fracturing of Si anodes during cycling.
In this review, we present recent advances in the development of advanced polymeric binders, including linear, branched, crosslinked, and self-healing polymers, to ensure high-performance Si-based anodes in LIBs.
2. Polymeric binders in LIBs
In LIBs, a polymeric binder serves an important role in the formation of an electrode by binding active materials and ensuring the adherence of the active materials on metallic current collectors such as copper or aluminum. Fig. 2(a) shows three possible binding mechanisms of a polymeric binder in electrodes. The first schematic illustrates the point binding mechanism, showing a uniform distribution of the binder and a good electronic conduction network. As depicted in Figs. 2(b) and (c), the second and third mechanisms are not desirable due to the poor electronic conduction by localized connections between the active materials and the electrical conducting agents, and due to an insulating polymer film which forms on the current collector. To ensure the electrochemical performance of electrodes, uniform binding by a polymeric binder is required to be maintained after the liquid electrolyte is absorbed into the binder matrix. Of course, the binder should be insoluble in a liquid electrolyte and should have good chemical stability against lithium salts and SEI components such as Li2CO3, LiF, and LiOH. More importantly, it was demonstrated that the mechanical strength of a binder in a dry state can be changed by the binder-electrolyte interaction [22,23].This means that the swellability of the binder in the electrode with the electrolyte affects the mechanical strength (binding ability and adhesion property) of the binder. It is noteworthy that undesirable access of the liquid electrolyte to the binder-Si interface weakens the mechanical strength of the binder. Therefore, to develop the appropriate binders for Si anodes, it is necessary that the mechanical strength of a binder in a wet state as well as in a dry state is considered.
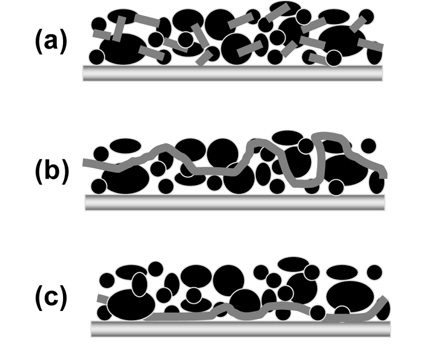
Schematic of three possible binding mechanisms of a polymeric binder in an electrode.
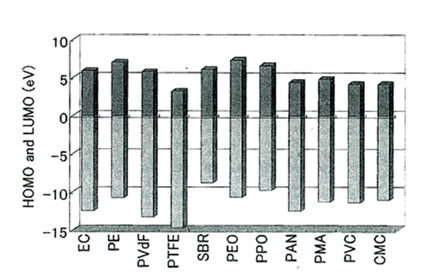
More importantly, for rational designs of binders, their electrochemical stability should be investigated. The primary means of discovering adequate binders is to calculate the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the polymers. The oxidation and reduction tendencies of the binders can be theoretically predicted through molecular orbital calculations, specifically ab initio quantum chemistry methods, which are computational chemistry methods based on quantum chemistry without experiments, as shown in Fig. 3. The bottom part of each bar represents the energy level of the HOMO of a certain molecule, and the top indicates the energy level of the LUMO of the same molecule. The LUMO of a polyethylene (PE) separator is relatively high compared to other materials, including the solvent of ethylene carbonate (EC). This implies that PE is electrochemically stable in a cathodic environment. Importantly, the HOMO of PE separator is higher than that of the EC solvent. It should be noted that a high HOMO energy level indicates that the compound is easy to oxidize in an anodic environment. In this regard, it is thought that the PE separator can scarcely remain stable at high voltages. On the other hand, the HOMO energies of poly(tetrafluoroethylene) (PTFE), PVdF (or PVDF), and polyacrylonitrile (PAN) are lower than those of other polymers. These polymers are expected to be electrochemically stable in a cathodic environment and to be very difficult to remove an electron from at high voltages. Therefore, these polymers can be used as a cathode binder. Importantly, the LUMO of PTFE is very low, as shown in Fig. 3. Because this polymer readily accepts electrons in a cathodic environment, it cannot be used as an anode binder. Unlike carbonaceous anodes with an acceptable degree of volume expansion (~10%), Si-based anodes undergo significant volume changes; thus, electrode deformation and external cell expansion take place. To alleviate the volume changes of Si-based anodes, mechanically robust and/or adhesive polymers have been studied as a binder because these polymers can accommodate severe volume changes and maintain a good electrical conduction network in the electrode.
As shown in Fig. 4, various polymer architectures of binders for Si-based anodes have been investigated. Poly(vinylene fluoride) (PVDF) is a linear-type homopolymer binder which is very commonly used in LIBs with graphite anodes. PVDF as a binder exhibits good binding ability to active materials/current collectors, reasonable electrochemical stability, and good wettability toward polar electrolyte solutions for facile Li ion transport. However, the PVDF binder does not endure large volume changes of Sibased anodes due to its insufficient mechanical strength [19]. To overcome the mechanical fracturing of electrodes induced by the extreme volumetric changes of Si, promising approaches to design appropriate polymer structures have been reported.
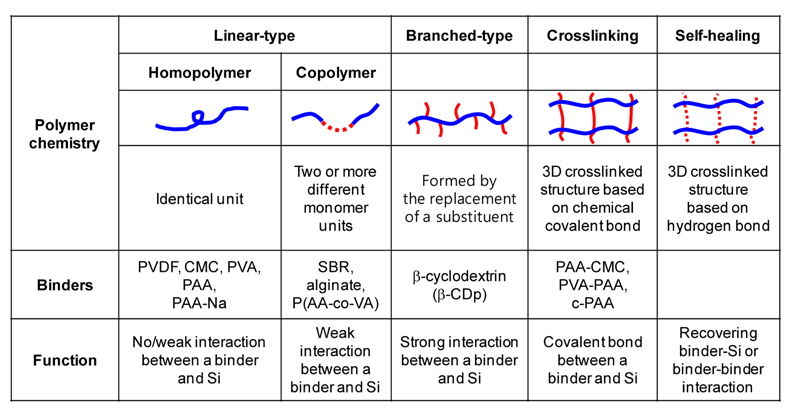
Polymer architectures for binders in Si-based anodes.
3. Linear-type polymeric binders
3.1. Homopolymers
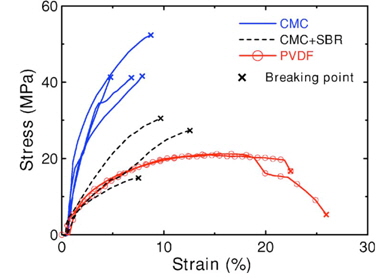
To improve the electrochemical performance of Si-based anodes, there are several strategies involving the use of a polymer with a simple linear structure. The Chen and Liu groups noted that the choice of binder is very important with regard to the cycling performance of alloy anodes [25-27]. The work of the Dahn group showed that the use of a water-soluble binder, carboxymethyl cellulose (CMC), could lead to an improvement in the electrochemical performance of commercially available Si (~325 mesh)-based anodes compared to a conventional PVDF binder [18]. This result suggests that brittle binders such as CMC may be able to endure the extreme volumetric changes of Si-based anodes. Although CMC film can be extended by only 5~8% before breaking, this type of CMC film has demonstrated relatively high mechanical strength compared to PVDF and CMC-SBR films, as shown in Fig. 5. The CMC binder may break during the Li insertion process.
Dahn et al. pointed out that CMC could play the additional role of modifying the surface chemistry of Si particles and thus reduce the capacity loss [18].It was reported that by varying levels of carboxymethyl (-OCH2COO−Na+) substitution, the adhesion of active materials to a copper current collector and the dispersion of the electrode slurry can be controlled [28]. Munao et al. demonstrated that the aggregation of PVDF binder and Si particles was more severe compared to a CMC-Si electrode; thus, the PVDF-Si electrode loses its electrical conduction network [29]. Although the Si-based anode with the CMC binder did not maintain its original morphological structure after the cycling test, the cycling stability of the Sibased anode was improved through the use of the CMC binder. Fig. 6 shows the binding mechanisms of the CMC binder with the Si nanoparticles [29]. Munao et al. noted that a weak bond formation between the CMC binder and Si nanoparticles by hydrogen bond interactions is a more favorable mechanism compared to the formation of a strong covalent bond between the -COOH of CMC and the -OH of the SiOx oxide layer covering the Si nanoparticles, as presented in Fig. 6 B. As also shown in Fig. 6, the formation of a hydrogen bond between the COOH of CMC and the OH of the nanoparticles can facilitate a self-healing process in Si-based anodes. Munao et al. demonstrated that this hydrogen bond interaction can break and reform in the presence of residual water [30].

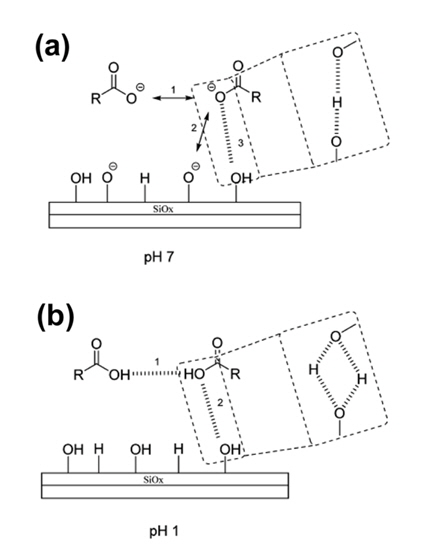
Recently, an interesting result related to the interaction between CMC and Si was reported by the Lux group [31]. At a pH of 7 of the slurry, there are three possible interactions for a CMC binder and the surface of the SiOx native oxide layer on Si nanoparticles, as shown in Fig. 7(a). First is a repulsive interaction between the deprotonated functional group (-RCO2−) of the CMC binder and another the deprotonated functional group of CMC (number 1 in Fig. 7(a)). Second is a repulsive interaction between the deprotonated functional group (-RCO2-) of the CMC binder and the negatively charged site on the Si surface (number 2 in Fig. 7(a)). Third is a hydrogen bonding interaction between the deprotonated functional group (-RCO2−) of CMC and the silanol (-OH) group of the SiOx native oxide layer on Si nanoparticles (number 3 in Fig. 7(a)). At a neutral pH value of the slurry, the more favorable interaction is the hydrogen bonding interaction between the CMC binder and the Si nanoparticle [31]. In an acidic environment (pH = 1), the functional moieties of CMC are protonated and the protonated groups on the Si surface are maintained. Thus, hydrogen bonding interactions of the CMC binder with another CMC and Si nanoparticles may occur at low pH values, as presented in Fig. 7(b). It can be considered that there is no preference for the two different interactions. From these mechanisms, it is likely that at high pH values, the dominant interaction is the hydrogen bonding interaction between the CMC and the Si nanoparticle.
Interestingly, Guyomard et al. reported that an esterification reaction between Si-OH on the Si surface and the COOH of the CMC binder preferentially occurred at pH = 3 and that this covalent bonding could remarkably improve the cycling performance of nano-Si anodes [32]. Oh et al. investigated a polyvinyl alcohol (PVA) linear-type polymer with many hydroxyl groups, showing that it can form hydrogen bonds with active materials and the current collector as a binder for Si/graphite composite anodes [33]. They found that the PVA binder with a high molecular weight improves the cycling performance of Si/graphite anodes compared to PVDF and polyacrylic acid (PAA) binders, as displayed in Fig. 8.

The work of the Yushin group showed that PAA with mechanical properties comparable to those of CMC and containing many carboxylic groups (-COOH) provides superior electrochemical performance as a polymeric binder for Si anodes [22]. However, Si anodes with Na-CMC or PVDF as a binder exhibited severe capacity fading during cycling. Interestingly, they explained that the use of a reducible additive, vinylene carbonate (VC) in this case, supports the long-term cycling stability of Si anodes. This indicates that a combination of a binder with functional additives which stabilize the anode surface is very promising approach for high-performance Si anodes. It is believed that hydrogen bonding between the car boxylic acid group of a polymeric binder such as CMC and the Si surface is highly effective at reducing the detrimental effects of a large volume expansion of Si anodes. Recently, carboxymethyl chitosan (C-chitosan), a water-soluble chitosan derivative prepared by the carboxymethylation of chitosan, was considered as a binder for Si anodes [34]. An Si anode with C-chitosan as a binder showed high initial Coulombic efficiency of 89% and delivered a high reversible capacity of 950 mAh g−1 at a current density of 500 mA g−1 over 50 cycles. The C-chitosan binder undergoes hydrogen bonding interaction with the Si surface, as shown in Fig. 9.

Fig. 10 shows the chemical structures of the PVDF, PAA-Na, and CMC-Na binders [35]. The reversible capacity of Si/graphite anodes was strongly dependent on the binder species at the initial cycle. The discharge (Li extraction) capacities were 400 and 800 mAh g−1 for the PVDF and CMC-Na binders, respectively. In contrast, the Si/graphite anode with the PAA-Na binder delivered a relatively high capacity of 1000 mAh g−1.

It was found that PAA-Na is the most appropriate polymeric binder to attain superior cycling stability of Si/graphite anodes (Fig. 11). Additionally, an increase in the amount of PAA-Na binder from 10 wt% to 30 wt% in the anode led to better cycling performance. Komaba et al. noted that the PAA-Na binder effectively suppressed the loss of the electrical conduction network in spite of the severe volumetric change of Si in the Si/graphite anode and thereby drastically improved the cycling performance.
3.2. Copolymers
In general, a conventional electrode is composed of active materials, a conducting agent such as carbon black, and a nonconductive polymeric binder. Metallic anode materials (Si, Sn, and Ge) undergo severe volume changes during Li+ insertion and extraction processes, breaking the electrical contact between the anode particles and the conductive additives. This mechanical fracture of negative electrodes (anodes) during cycling results in the isolation of anode particles from the electrical conducting network in the anode and degradation of the electrodes. Xun et al. demonstrated that a conductive polymeric binder performs the unique function of maintaining the electronic integrity of the anode, as depicted in Figs. 12(a) and (b) [36].Conductive additives do not maintain their electrical connectivity in a metallic anode which undergoes significant volume expansion during Li insertion, whereas poly(9,9-dioctylfluorene-co-fluorenone-co-methylbenzoic ester) (PFM) with two functional groups, carbonyl and methylbenzoic ester, as a conductive binder provides not only good mechanical binding force but also electrical integrity. The Liu group attempted to design a polymeric binder which provided good electrical conductivity, mechanical adhesion and flexibility with inexhaust ible tolerance against large volume changes, as well as good compatibility with the electrolyte solution for high-capacity metallic anodes [37].
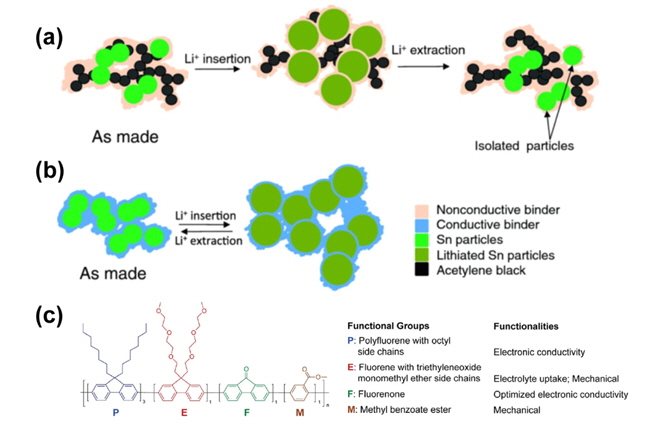
Fig. 12(c) shows the chemical structure of poly(2,7-9,9-dioctylfluorene-co-2,7-9,9-(di(oxy-2,5,8-trioxadecane))fluorene-co-2,7-fluorenone-co-2,5-1-methylbenzoate ester) (PEFM), which can maintain high electrical conductivity (fluorene with octyl side chains), mechanical adhesion (fluorene with ethylene oxide side chains and methylbenzoate ester unit), flexibility, and appropriate electrolyte uptake (ethylene oxide side chains) to ensure proper ionic conductivity.
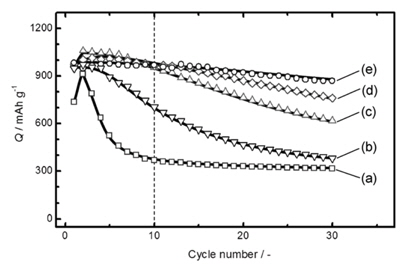
To understand the effect of a conductive polymeric binder on the cycling performance of Si electrodes, the Liu group fabricated conductive polymer/Si electrodes without conductive additives. The PEFM conductive polymer in their study retained a high Li extraction capacity of 3000 mAh g−1 at room temperature for more than 50 cycles (Fig. 13(a)). Superior capacity retention of Si was observed in the PEFM as compared to the PFM binder. This result indicates that the utilization of Si embedded in the PEFM binder is much higher than that in the PFM binder. The PFM binder that does not contain a fluorene unit and contains a triethylene oxide monomethyl ether side chain, which exhibits a relatively low Li extraction capacity at a rate of C/10, while the Si anode with the PEFM conductive binder delivered nearly full theoretical capacity (~3500 mAh g−1), as shown in Fig. 13(b). In addition, the Si anode with the PEFM conductive binder showed superior Li extraction capacity at a high rate of 2 C compared to the PFM binder (Fig. 13(b)). This indicates that the bridges between the PEFM conductive binder and the Si particles are robust enough to preserve the electrical conductive network and facilitate the electro-chemical reaction of Li with Si during the insertion and extraction of Li at high rates.
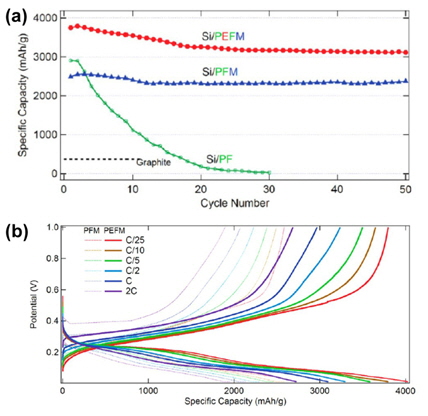
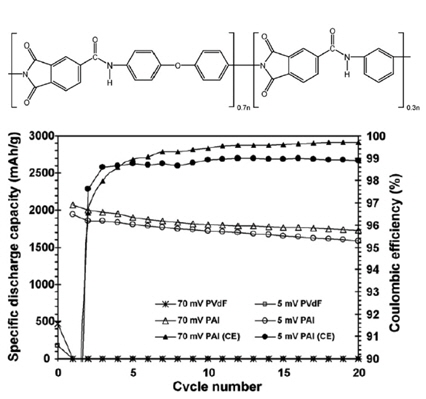
In an attempt to overcome the mechanical disintegration of a Si-based anode due to large changes in the volume during Li+ insertion and extraction, our group found that the mechanical properties of polymeric binders are critical factors in the colossal volume expansion of Si micro-sized particles [19]. Indeed, polyamide imide (PAI) with a high mechanical strength drastically improved the electrochemical performance of micro-Si anodes compared to a commercial PVDF binder. Although the PAI binder electrochemically reacted with Li ions and electrons during the first charge process, the cyclic degradation of the active Si material could be remarkably reduced by means of the PAI binder given its high tensile strength.
Recently, a binder that has a positive effect on the electrochemical performance of Si-based electrodes was reported [23]. Specifically, a high-modulus natural polysaccharide extracted from brown algae was found to yield a substantially stable Si-based anode compared with polymeric binders such as PVDF, PAA, and CMC. Unlike the polysaccharides commonly found in plants, alginates (alginic acid, commonly in a Na salt form) of brown algae have a high content of carboxyl moiety, which can lead to many possible bonds with Si particles. Importantly, the carboxyl moieties in Na alginate are uniformly distributed, unlike CMC with a random distribution of carboxyl groups. It was explained that this uniform distribution of carboxyl moieties can lead to a more uniform coverage and more efficient assistance with the formation of SEI on the Si. The Yushin group mentioned that the alginate used in their work is a copolymer of b-D-mannuronic acid and a-L-guluronic acid (Fig. 15(a)) and that the ratio of M-to-G monoblocks in the alginates is 1.13. The Na alginate exhibited greater stiffness than PVDF films by about 6.7 times in a dry state. This mechanical characteristic may help mitigate the large volume expansion of Si. They described that the Na alginate binder provides appropriate binder-electrolyte interactions and facile access of Li ions to the Si surface and that it assists in the creation of a stable solid-electrolyte interphase (SEI) on the Si surface [23]. As shown in Fig. 15(b), an alginate binder allowed for stable cycling performance of nano-Si anodes compared to PVDF and Na-CMC binders. Although both CMC and Na alginate show similar mechanical properties, they suggested that the inferior cycling performance of Si-CMC electrodes stems from the random distribution of carboxyl groups in the CMC binder. In spite of the superior performance of Na alginate, its hydroxyl groups remaining after vacuum annealing at 105℃ are a critical problem; they may react with the PF5 produced from the LiPF6 salt at elevated temperatures [38], and the resulting phosphorous oxyfluoride (OPF3) will lead to the decomposition of carbonate solvents and result in severe degradation of the Si anodes.

(a) Chemical structure of mannuronic (left) and guluronic (right) acids. (b) Li-extraction capacity of nano-Si anodes with alginate, CMC, and PVDF binders. The current density was 4200 mA/g (corresponding to a rate of 1C) and Li/Si half cells were cycled in the potential window of 0.01 to 1 V versus Li/Li+ [23].
Recently, mussel-inspired adhesive materials with catechol functional group were investigated as a binder. The catechol moiety with wetness-resistance adhesion in a liquid environment was introduced into PAA and CMC as a side chain, as presented in Fig. 16(a) [39].
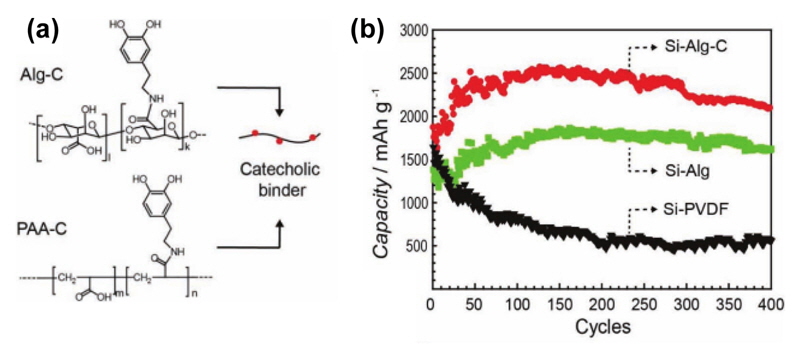
The addition of catechol groups into the alginate binder delivered high Li extraction capacity compared to an alginate binder without catechol groups (Fig. 16(b)). This result indicates that catechol groups preserve the electronic network well during cycling and allow reversible electrochemical reactions of the Si anode.
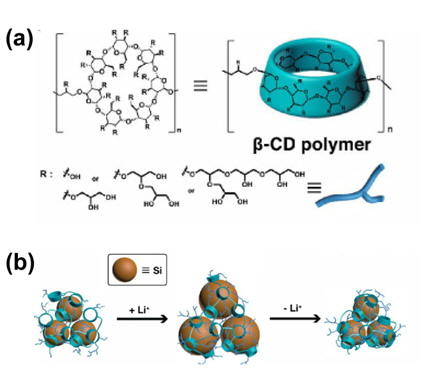
The Choi group found that linear-type polymers have limited interactions with nano-Si particles. To improve the interactions between nano-Si particles and a binder, they proposed hyperbranched polymeric binders with an increased number of contacts between the nano-Si particles and the binders, as shown in Fig. 17(a) [40]. A β-CDp binder with a hyperbranched main backbone creates a fractal-like structure-based network in the Si anode; its various bulky side groups are physically entangled with each other via hydrogen bonding and van der Waals force, as displayed in Fig. 17(b). It was reported that this polymeric network created by the β-CDp binder accommodates a large volume expansion of Si during Li insertion and assists in the recovery of the interactions with Si after it loses contact with the binder at certain points during the Li extraction processs [40]. In addition, a further improvement of the electro-chemical performance of Si anodes could be realized with a hybrid of the hyperbranched β-CDp binder and a one-dimensional (D) alginate binder. They noted that the introduction of the 1D alginate binder into β-CDp reduces the agglomeration of β-CDp and leads to a more uniformly distributed polymeric network through the neutralization of electrostatic repulsion between the carboxylate groups in 1D alginate by β-CDp [40].
4. Crosslinked polymeric binders
Our group reported a thermally cured polymeric binder as an immediate technological solution for Liion batteries with Si-based electrodes [7]. A three-dimensionally interconnected network of PAA and sodium carboxymethyl cellulose (Na-CMC) as a binder was used and a nano-sized silicon anode with this crosslinked binder displayed a high Li extraction capacity that exceeded 2000 mAh g−1 after 100 cycles at 30℃. The crosslinked polymeric binder (Fig. 4) showed high mechanical resistance to strain, particularly non-recoverable deformation because the polymer chains are three-dimensionally linked. This was the first report which showed chemical crosslinking between two different polymers for Si anodes, after which various three-dimensionally crosslinked polymeric binders were proposed.
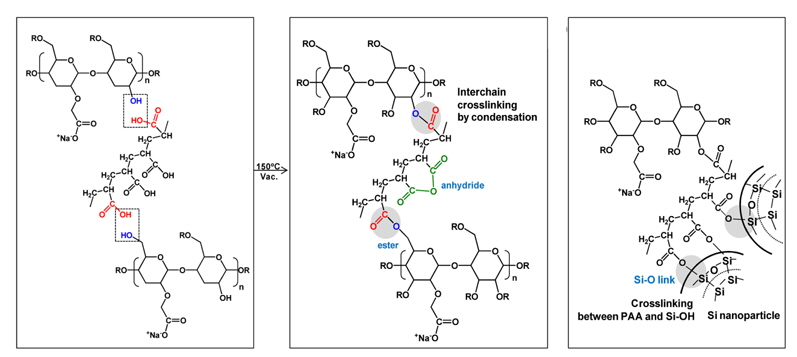
In order to enhance the mechanical properties of a polymeric binder, a photo-crosslinked polymer system was proposed [41]. In that study, a robust PAA polymer (PAA-BP) which was functionalized with photo-cross-linkable benzophenone (BP) as a binder for Si-based anodes was synthesized, as shown in Fig. 19(a). The Si anode with the crosslinked PAABP binder exhibited lower deformation upon a volume change. It was noted that the three-dimensionally interconnected structure allows good electrical contact between the Si nanoparticles and an electrical conducting additive (carbon black) in the electrode [42]. This result was supported by ex-situ thickness measurements of Si anodes after 60 cycles (Figs. 19(b) and (c)). The non-crosslinked PAA-BP binder showed a more expanded Si anode compared to the crosslinked PAA-BP. This indicates that the crosslinked PAA-BP binder effectively accommodates the volume changes of Si anodes upon lithiation and delithiation.
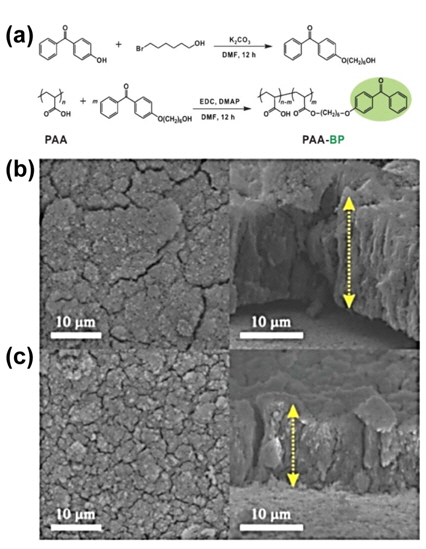
The Cui group examined an in-situ polymerized conducting hydrogel system for the conformal coating of the binder onto Si nanoparticles, as displayed in Fig. 20(a) [42]. They demonstrated that the resulting hierarchical hydrogel framework provides a continuous electrically conductive polyaniline (PANi) network, binding with the Si surface via crosslinker hydrogen bonding with phytic acid or electrostatic interaction between the positively charged PANi doped with phytic acid and the negatively charged surface oxide (-Si-O-) on the Si, with a porous space for a large volume expansion of the Si nanoparticles. The Si nanoparticle-PANi hydrogel composite is fabricated by solution-phase synthesis by mixing Si nanoparticles with phytic acid and an aniline monomer in water to produce a suspension [43]. The Si-PANi composite electrode exhibited relatively stable cycling performance compared to other samples and delivered a high Li extraction capacity of ~1,200 mAh g−1 after 1,000 cycles (Fig. 20(b)).
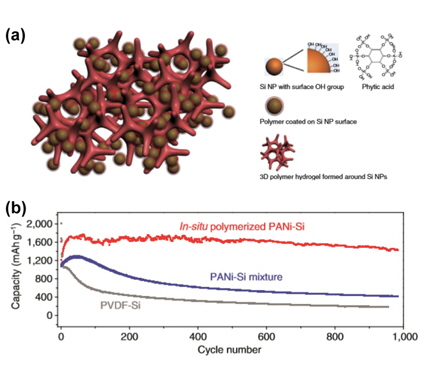
(a) Schematic of 3D porous Si nanoparticle (NP)/conductive PANi polymer hydrogel composite electrodes. Each Si NP is covered with a conductive PANi polymer surface coating and is further connected to a highly porous hydrogel framework. (b) Comparison of the Li extraction capacity retention behaviors of in-situ polymerized SiNP-PANi composite electrodes (red line), the PANi-Si mixture (blue line), and PVDF-Si (grey line) for a charge/discharge current density of 1.0 Ag−1 between 0.01 V and 1.0 V versus Li/Li+ [42].
Komaba et al. reported crosslinked PAA with polycarbodiimide (PCD) as a binder for a Si/graphite composite anode [43]. They demonstrated the positive effect of PCD as a crosslinker for a PAA binder on the electrochemical performance of Si/graphite anodes. PCD was suggested to modulate the polymer conformation and rheological properties of the slurry in a NMP solvent. This cross-link process also improves the mechanical stability of the composite electrode against the strain induced by the severe volume expansion of Si nanoparticles upon Li insertion. The Si/graphite anode with the crosslinked PAA-1% PCD delivered more than 1,000 mAh g−1 and showed improved cycling stability compared to PVDF and PAA binders [43]. Recently, Ryu et al. synthesized polymers with carboxylic acid (COOH), carboxylate (COO-), and hydroxyl (OH) groups in a single polymer backbone to develop a new polymeric binder system for Si anodes in LIBs [44]. This synthesized poly(acrylic acid-co-vinyl alcohol) random copolymer could lead to the formation of a three-dimensionally interconnected network via ester formation between carboxylic acid and alcohol during the solvent-drying process for the preparation of the electrode. The resulting crosslinked binder led to superior electrochemical performance of a nano-sized Si anode.
Zheng et al. proposed a facile strategy to construct a 3D polymeric network as a binder for Si sub-microparticle-based anodes [45]. The 3D polymeric network with a high degree of crosslinking could improve the mechanical properties of Si-based anodes through interconnecting alginate chains via a high concentration of Ca2+ cations and forming strong hydrogen bonds between the free carboxylic groups of alginate and the hydroxylated Si surface, as depicted in Fig. 21(a). The hydrogen bonding between the free carboxylic groups of alginate and the hydroxylated Si surface is considered as one of the most important parameters affecting the effective anchoring of the binder onto the Si surface with a self-healing process (the breaking and reformation of the bonds upon lithiation and delithiation) [37,46,47].
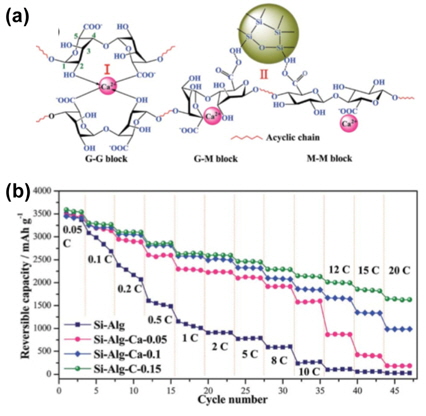
(a) Schematic drawing of the robust coordinate bonds between alginate chains and calcium cations (regionand strong hydrogen bonding between a hydroxylated Si surface and free alginate carboxylic groups (region II). (b) Rate capabilities of Si sub-microparticle (~200 nm)-alginate laminates with a 3D alginate network with dierent degrees of crosslinking [45].
Fig. 21(b) shows the rate capabilities of Si-based anodes with alginate of various calcium molar fractions. The Si-Alg-Ca-0.15 anodes exhibited superior rate capabilities at high C rates compared to the other samples. This was noted as a drastic improvement in the rate capability of the Si anodes due to the high mechanical properties of the crosslinked alginate binder.
Very recently, a crosslinked polymeric system very similar to that in one of our previous reports was explored as an interpenetrated gel polymer binder for Si anodes, as shown in Fig. 22(a) [48]. The researchers in that study prepared the Si anodes by means of the in-situ thermal crosslinking of water-soluble PAA and PVA polymers. A comparison of the cycling performances of Si anodes with PVDF, NaCMC, and PAA-PVA binders at a current density of 400 mA g−1 is shown in Fig. 22(b).
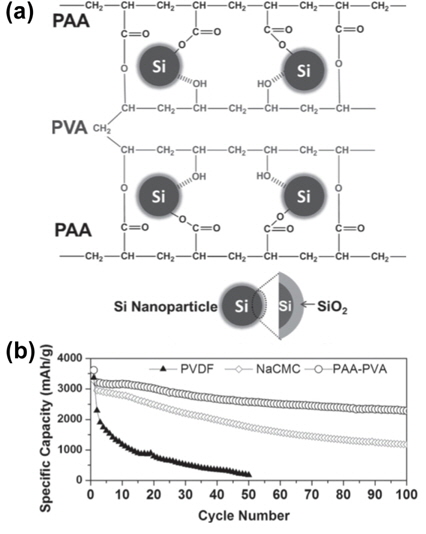
The cell with the PVDF binder showed severe capacity fading (only 180 mAh g−1 after 50 cycles), while Si anodes with the NaCMC and PAA-PVA binders exhibited improved cycling stability. Specifically, the Si anode with the PAA-PVA binder delivered a very high Li extraction capacity of 3616 mAh g−1 during the initial cycling, which is nearly 86% of the theoretical capacity (4200 mAh g−1). It also demonstrated superior cycling stability, with a capacity of 2283 mAh g−1 after 100 cycles (Fig. 22(b)).
5. Self-healing polymeric binders
The Bao group proposed self-healing chemistry to repair mechanically damaged Si-based electrodes spontaneously [49]. They noted that the crosslinked network of self-healing polymers could be stretched to three times its initial length without breaking, whereas PVDF and alginate binders could be stretched only by 10% and 2%, respectively. As shown in Fig. 23(a), it was predicted that the self-healing polymer would be stretchable and repair spontaneously the mechanical fracturing of Si-based anodes via hydrogen bonding. The cycling stability and rate capability of Si microparticle anodes were greatly improved [49]. The self-healing Si anodes retained a high Li extraction capacity of ~2,500 mAh g−1 during 20 cycles.

The Coskun group reported a functional binder with Meldrum’s acid for Si anodes, as presented in Fig. 24(a). Their binder could lead to no/weak interactions between polymer chains and Si particles, reactions with the native silanol groups on the Si surface, and covalent crosslinking. Here, “self-healing” refers to the capability to recover binder-Si or binder-binder interactions in situations that may cause a loss of such interactions due to the large volume changes of Si-active materials, as depicted in Fig. 24(a).
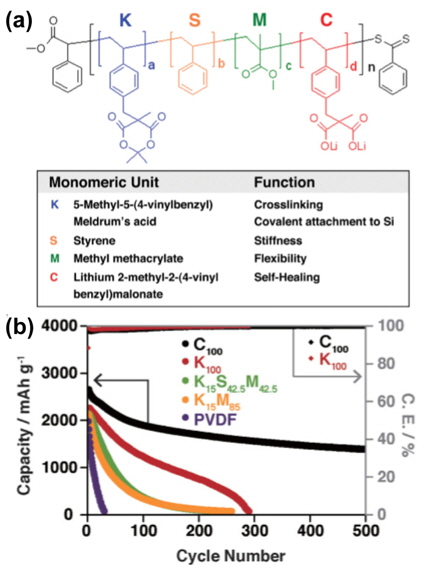
The Coskun group attempted to identify the most important factor pertaining to a binder for Si anodes. The cycling performance of Si anodes was comparatively investigated while focusing on the various chemical properties of binders, i.e., crosslinking, covalent attachment, stiffness, exibility, and self-healing. First, the PVDF binder showed the worst cycling properties, with only weak van der Waals interactions with Si (Fig. 24(b)). The Li extraction capacity retention of a K15S42.5M42.5-containing Si anode was slightly improved compared to K15M85 15 M 85, which suggests that the stiffness introduced by the S unit provides a minor improvement in the cycling performance. Importantly, the cycling performance of K100 was greatly improved compared to that of KM and KSM, indicating that the crosslinking/covalent attachment is more critical than the stiffness of the individual polymer chains for improving the cycling performance.
6. Challenges and perspective
We demonstrated various polymeric binder systems as promising approaches for the development of commercially viable Si-based anodes for Li-ion batteries with high energy densities. In general, a polymeric binder should provide the proper viscosity of the slurry to ensure the uniform dispersion of the active materials and the electrically conducting additives, while also facilitating good chemical and electrochemical stability against the cell components. In addition to these requirements, a binder should accommodate a large volume expansion (ca. 400%) of Si anode materials upon Li insertion to inhibit the mechanical fracture of Si anodes during cycling. It was clearly described that the mechanical properties of the binder have a significant impact on the Si-based anode materials when they undergo significant volume changes during cycling. Among the various approaches taken to realize high-performance Si-based anodes, one of the most promising strategies is to develop mechanically robust binders that can inhibit severe volume changes. When polymer chains are linked together by crosslinking, they lose some of their ability to move as individual polymer chains and create a mechanically robust polymer matrix. The resulting three-dimensionally interconnected polymeric binder could effectively alleviate the severe volume expansion of Si anode materials during the Li insertion process. Self-healing polymers based on a three-dimensionally interconnected network are regarded as a highly promising means to accommodate the large volume changes of Si-based anodes. Importantly, using electrolyte additives to build-up a thin and stable artificial SEI layer as well as Si-active materials with an optimized structure should be considered to ensure the long-term cycling stability of Si-based electrodes. In addition, the existence of an exterior carbon or metal (metal oxide) shell will support freedom of expansion, improve the electrical contact, and serve as a physical barrier to electrolyte decomposition.
Acknowledgements
This research was supported by the IT R&D program of MKE/KEIT (KI001810039182, development of 5V cathode material which capacity is 125 mAh g−1 and high voltage electrolyte which decomposition is over 5V for lithium secondary battery) and by a grant from the Energy Efficiency & Resources of the Korea Institute of Energy Technology Evaluation and Planning (Project no. 20112010100140) funded by the Korean Ministry of Knowledge Economy. Part of this research was funded by Samsung SDI.
References
L.W. Su, Y. Jing, Z. Zhou, Nanoscale, 10, 3967(2011).
Su L.W., Jing Y., Zhou Z.. Nanoscale 2011;10:3967.M. Hu, X.L. Pang, Z. Zhou, J. Power Sources, 237, 229(2013).
Hu M., Pang X.L., Zhou Z.. J. Power Sources 2013;237:229. 10.1016/j.jpowsour.2013.03.024.J.-M. Tarascon, M. Armand, Nature, 414, 359(2001).
Tarascon J.-M., Armand M.. Nature 2001;414:359. 10.1038/35104644.N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho, P. G. Bruce, Angew. Chem. Inter. Ed., 51, 9994(2012).
Choi N.-S., Chen Z., Freunberger S. A., Ji X., Sun Y.-K., Amine K., Yushin G., Nazar L. F., Cho J., Bruce P. G.. Angew. Chem. Inter. Ed. 2012;51:9994. 10.1002/anie.201201429.B. Dunn, H. Kamath, J.-M. Tarascon, Science, 334, 928(2011).
Dunn B., Kamath H., Tarascon J.-M.. Science 2011;334:928. 10.1126/science.1212741.C. K. Chan, H. Peng, G. Liu, K. Mcllwrath, X. F. Zhang, R. A. Huggins, Y. Cui, Nat. Nanotechnol., 3, 31(2008).
Chan C. K., Peng H., Liu G., Mcllwrath K., Zhang X. F., Huggins R. A., Cui Y.. Nat. Nanotechnol. 2008;3:31. 10.1038/nnano.2007.411.B. Koo, H. Kim, Y. Cho, K.T. Lee, N.-S. Choi, J. Cho, Angew. Chem. Int. Ed., 51, 8762(2012).
Koo B., Kim H., Cho Y., Lee K.T., Choi N.-S., Cho J.. Angew. Chem. Int. Ed. 2012;51:8762. 10.1002/anie.201201568.Y.-S. Hu, R. Demir-Cakan, M.-M. Titirici, J.-O. Müller, R. Schlögl, M. Antonietti, J. Maier, Angew. Chem. Int. Ed., 47, 1645(2008).
Hu Y.-S., Demir-Cakan R., Titirici M.-M., Müller J.-O., Schlögl R., Antonietti M., Maier J.. Angew. Chem. Int. Ed. 2008;47:1645. 10.1002/anie.200704287.J.I. Lee, N.-S. Choi, S. Park, Energy & Environ. Sci., 5, 7878(2012).
Lee J.I., Choi N.-S., Park S.. Energy & Environ. Sci. 2012;5:7878. 10.1039/c2ee21380j.G. Jeong, S.M. Lee, N.-S. Choi, Y.U. Kim, C.K. Lee, Electrochim. Acta, 56, 5095(2011).
Jeong G., Lee S.M., Choi N.-S., Kim Y.U., Lee C.K.. Electrochim. Acta 2011;56:5095. 10.1016/j.electacta.2011.03.071.N.-S. Choi, Y. Yao, Y. Cui, J. Cho, J. Mater. Chem., 21, 9825(2011).
Choi N.-S., Yao Y., Cui Y., Cho J.. J. Mater. Chem. 2011;21:9825. 10.1039/c0jm03842c.H. Wu, Y. Cui, Nano Today, 7, 414(2012).
Wu H., Cui Y.. Nano Today 2012;7:414. 10.1016/j.nantod.2012.08.004.M. Yoshio, H. Wang, K. Fukuda, T. Umeno, N. Dimov, Z. Ogumi., J. Electrochem. Soc., 149, A1598(2002).
Yoshio M., Wang H., Fukuda K., Umeno T., Dimov N., Ogumi Z.. J. Electrochem. Soc. 2002;149:A1598. 10.1149/1.1518988.T.D. Hatchard, J.R. Dahn, J. Electrochem. Soc., 151, A1628(2004).
Hatchard T.D., Dahn J.R.. J. Electrochem. Soc. 2004;151:A1628. 10.1149/1.1790533.H. Kim, B. Han, J. Choo, J. Cho, Angew. Chem. Int. Ed., 47, 10151(2008).
Kim H., Han B., Choo J., Cho J.. Angew. Chem. Int. Ed. 2008;47:10151. 10.1002/anie.200804355.J. Cho, J. Mater. Chem., 20, 4009(2010).
Cho J.. J. Mater. Chem. 2010;20:4009. 10.1039/b923002e.D. Guy, B. Lestriez, D. Guyomard, Adv. Mater., 16, 553(2004).
Guy D., Lestriez B., Guyomard D.. Adv. Mater. 2004;16:553. 10.1002/adma.200306075.J. Li, R.B. Lewis, J.R. Dahn, Electrochem. Solid-State Lett., 10, A17(2007).
Li J., Lewis R.B., Dahn J.R.. Electrochem. Solid-State Lett. 2007;10:A17. 10.1149/1.2398725.N.-S. Choi, K.H. Yew, W.-U. Choi, S.-S. Kim, J. Power Sources, 177, 590(2008).
Choi N.-S., Yew K.H., Choi W.-U., Kim S.-S.. J. Power Sources 2008;177:590. 10.1016/j.jpowsour.2007.11.082.A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C.F. Huebner, T.F. Fuller, I. Luzinov, G. Yushin, ACS Appl. Mater. & Inter., 2, 3004(2010).
Magasinski A., Zdyrko B., Kovalenko I., Hertzberg B., Burtovyy R., Huebner C.F., Fuller T.F., Luzinov I., Yushin G.. ACS Appl. Mater. & Inter. 2010;2:3004. 10.1021/am100871y.N.S. Hochgatterer, M.R. Schweiger, S. Koller, P.R. Raimann, T. Wöhrle, C. Wurm, M. Winter, Electrochem. Solid-State Lett., 11, A76(2008).
Hochgatterer N.S., Schweiger M.R., Koller S., Raimann P.R., Wöhrle T., Wurm C., Winter M.. Electrochem. Solid-State Lett. 2008;11:A76. 10.1149/1.2888173.A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C.F. Huebner, T.F. Fuller, I. Luzinov, G. Yushin, ACS Applied Mater. & Inter., 2, 3004(2010).
Magasinski A., Zdyrko B., Kovalenko I., Hertzberg B., Burtovyy R., Huebner C.F., Fuller T.F., Luzinov I., Yushin G.. ACS Applied Mater. & Inter. 2010;2:3004. 10.1021/am100871y.I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov, G. Yushin, Science, 334, 75(2011).
Kovalenko I., Zdyrko B., Magasinski A., Hertzberg B., Milicev Z., Burtovyy R., Luzinov I., Yushin G.. Science 2011;334:75. 10.1126/science.1209150.K. Kiyoshi, Japan CMC Press, 2003
. Kiyoshi K.. Japan CMC Press; 2003.Z. Chen, L. Christensen, J.R. Dahn, Electrochem. Commun., 5, 919(2003).
Chen Z., Christensen L., Dahn J.R.. Electrochem. Commun. 2003;5:919. 10.1016/j.elecom.2003.08.017.W.R. Liu, M.H. Yang, H.C. Wu, S.M. Chiao, N.L. Wua, Electrochem. Solid-State Lett., 8, A100(2005).
Liu W.R., Yang M.H., Wu H.C., Chiao S.M., Wua N.L.. Electrochem. Solid-State Lett. 2005;8:A100. 10.1149/1.1847685.Z. Chen, L. Christensen, J.R. Dahn, J. Electrochem. Soc., 150, A1073(2003).
Chen Z., Christensen L., Dahn J.R.. J. Electrochem. Soc. 2003;150:A1073. 10.1149/1.1586922.J.-H. Lee, U. Paik, V.A. Hackney, Y.-M. Choi, J. Electrochem. Soc., 152, A1763(2005).
Lee J.-H., Paik U., Hackney V.A., Choi Y.-M.. J. Electrochem. Soc. 2005;152:A1763. 10.1149/1.1979214.D. Munao, J.W.M. Van-Erven, M. Valvo, E. Garcia-Tamayo, E.M. Kelder, J. Power Sources, 196, 6695(2011).
Munao D., Van-Erven J.W.M., Valvo M., Garcia-Tamayo E., Kelder E.M.. J. Power Sources 2011;196:6695. 10.1016/j.jpowsour.2010.11.072.P. Cordier, F. Tournilhac, C. Soulié-Ziakovic, L. Leibler, Nature, 451, 977(2008).
Cordier P., Tournilhac F., Soulié-Ziakovic C., Leibler L.. Nature 2008;451:977. 10.1038/nature06669.U.S. Vogl, P.K. Das, A.Z. Weber, M. Winter, R. Kostecki, S.F. Lux, Lanmuir, 30, 10299(2014).
Vogl U.S., Das P.K., Weber A.Z., Winter M., Kostecki R., Lux S.F.. Lanmuir 2014;30:10299. 10.1021/la501791q.D. Mazouzi, B. Lestriez, L. Roué, D. Guyomard, J. Electrochem. Soc., 12, A215(2009).
Mazouzi D., Lestriez B., Roué L., Guyomard D.. J. Electrochem. Soc. 2009;12:A215.H.-K. Park, B.-S. Kong, E.-S. Oh, Electrochem. Commun., 13, 1051(2011).
Park H.-K., Kong B.-S., Oh E.-S.. Electrochem. Commun. 2011;13:1051. 10.1016/j.elecom.2011.06.034.L. Yue, L. Zhang, H. Zhong, J. Power Sources, 247, 327(2014).
Yue L., Zhang L., Zhong H.. J. Power Sources 2014;247:327. 10.1016/j.jpowsour.2013.08.073.S. Komaba, N. Yabuuchi, T. Ozeki, Z.-J. Han, K. Shimomura, H. Yui, J. Phys. Chem. C, 116, 1380(2012).
Komaba S., Yabuuchi N., Ozeki T., Han Z.-J., Shimomura K., Yui H.. J. Phys. Chem. C 2012;116:1380. 10.1021/jp204817h.S. Xun, X. Song, V. Battaglia, G. Liu, J. Electrochem. Soc., 160, A849, 2013.
Xun S., Song X., Battaglia V., Liu G.. J. Electrochem. Soc. 2013;160:A849–849.M. Wu, X. Xiao, N. Vukmirovic, S. Xun, P.K. Das, X. Song, P. Olalde-Velasco, D. Wang, A.Z. Weber, L.-W. Wang, V.S. Battaglia, W. Yang, G. Liu, J. Am. Chem. Soc., 135, 12048(2013).
Wu M., Xiao X., Vukmirovic N., Xun S., Das P.K., Song X., Olalde-Velasco P., Wang D., Weber A.Z., Wang L.-W., Battaglia V.S., Yang W., Liu G.. J. Am. Chem. Soc. 2013;135:12048. 10.1021/ja4054465.C.L. Campion, W. Li, B.L. Lucht, J. Electrochem. Soc., 152, A2327(2005).
Campion C.L., Li W., Lucht B.L.. J. Electrochem. Soc. 2005;152:A2327. 10.1149/1.2083267.M.-H. Ryou, J. Kim, I. Lee, S. Kim, Y.K. Jeong, S. Hong, J.H. Ryu, T.-S. Kim, J.-K. Park, H. Lee, J.W. Choi, Adv. Mater., 25, 1571(2013).
Ryou M.-H., Kim J., Lee I., Kim S., Jeong Y.K., Hong S., Ryu J.H., Kim T.-S., Park J.-K., Lee H., Choi J.W.. Adv. Mater. 2013;25:1571. 10.1002/adma.201203981.Y.K. Jeong, T.-W. Kwon, I. Lee, T.-S. Kim, A. Coskun, J.W. Choi, Nano Lett., 14, 864(2014).
Jeong Y.K., Kwon T.-W., Lee I., Kim T.-S., Coskun A., Choi J.W.. Nano Lett. 2014;14:864. 10.1021/nl404237j.Y. Park, S. Lee, S.-H. Kim, B.Y. Jang, J.S. Kim, S.M. Oh, J.-Y. Kim, N.-S. Choi, K.T. Lee, B.-S. Kim, RSC Advances, 3, 12625(2013).
Park Y., Lee S., Kim S.-H., Jang B.Y., Kim J.S., Oh S.M., Kim J.-Y., Choi N.-S., Lee K.T., Kim B.-S.. RSC Advances 2013;3:12625. 10.1039/c3ra42447b.H. Wu, G. Yu, L. Pan, N. Liu, M.T. McDowell, Z. Bao, Y. Cui, Nature Commun., 4,1943(2013).
Wu H., Yu G., Pan L., Liu N., McDowell M.T., Bao Z., Cui Y.. Nature Commun. 2013;4:1943.Z.-J. Han, N. Yabuuchi, S. Hashimoto, T. Sasaki, S. Komaba, ECS Electrochem. Lett., 2, A17(2013).
Han Z.-J., Yabuuchi N., Hashimoto S., Sasaki T., Komaba S.. ECS Electrochem. Lett. 2013;2:A17. 10.1149/2.003309eel.M.T. Jeena, J.-I. Lee, S.H. Kim, C. Kim, J.-Y. Kim, S. Park, J.-H. Ryu, ACS Appl. Mater. & Inter., 6, 18001(2014).
Jeena M.T., Lee J.-I., Kim S.H., Kim C., Kim J.-Y., Park S., Ryu J.-H.. ACS Appl. Mater. & Inter. 2014;6:18001. 10.1021/am504854x.L. Zhang, L. Zhang, L. Chai, P. Xue, W. Hao, H. Zheng, J. Mater. Chem. A, 2, 19036(2014).
Zhang L., Zhang L., Chai L., Xue P., Hao W., Zheng H.. J. Mater. Chem. A 2014;2:19036. 10.1039/C4TA04320K.J.-S. Bridel, T. Azaïs, M. Morcrette, J.-M. Tarascon, D. Larcher, Chem. Mater., 22, 1229(2010).
Bridel J.-S., Azaïs T., Morcrette M., Tarascon J.-M., Larcher D.. Chem. Mater. 2010;22:1229. 10.1021/cm902688w.G. Liu, S, Xun, N, Vukmirovic, X, Song, P. Olalde-Velasco,H. Zheng, V.S. Battaglia, L. Wang, W. Yang, Adv. Mater., 23, 4679(2011).
Liu G., Xun S,, Vukmirovic N,, Song X,, Olalde-Velasco P., Zheng H., Battaglia V.S., Wang L., Yang W.. Adv. Mater. 2011;23:4679. 10.1002/adma.201102421.J. Song, M. Zhou, R. Yi, T. Xu, M.L. Gordin, D. Tang, Z. Yu, M. Regula, D. Wang, Adv. Funct. Mater., 24,5904(2014).
Song J., Zhou M., Yi R., Xu T., Gordin M.L., Tang D., Yu Z., Regula M., Wang D.. Adv. Funct. Mater. 2014;24:5904. 10.1002/adfm.201401269.C. Wang, H. Wu, Z. Chen, M.T. McDowell, Y. Cui, Z. Bao, Nature Chem., 5, 1042(2013).
Wang C., Wu H., Chen Z., McDowell M.T., Cui Y., Bao Z.. Nature Chem. 2013;5:1042. 10.1038/nchem.1802.T.-W. Kwon, Y.K. Jeong, I. Lee, T.-S. Kim, J.W. Choi, A. Coskun, Adv. Mater., 26, 7979(2014).
Kwon T.-W., Jeong Y.K., Lee I., Kim T.-S., Choi J.W., Coskun A.. Adv. Mater. 2014;26:7979. 10.1002/adma.201402950.