1. Introduction
With the progression of climate change and global warming, the development of alternative energy for reducing the use of fossil fuels is an inevitable and crucially important undertaking. Reverse electrodialysis (RED) is receiving growing attention as a sustainable energy technology for generating electric power from salinity gradients without thermal pollution or CO2 emission [1ŌĆō3]. Membrane potential is established from alternating anion exchange membranes (AEMs) and cation exchange membranes (CEMs) and diffusion transport of ions is converted to electric current by redox reaction at the electrodes. The theoretical energy generated from RED is estimated to be 1.7 MJ when 1 m3 sea water is mixed with the same amount of river water. Unlike intermittent solar and wind energy, RED can be continuously available for conversion into electric power for 24 h.
Since Pattle [4] first demonstrated a membrane stack for electric power in the early 1950ŌĆÖs and Weinstein [5] emphasized engineering, economic, and environmental considerations that must be resolved in the 1970ŌĆÖs, there have been significant achievements [6] in development of RED components [7ŌĆō12] (e.g., membranes and spacers) and extensive analysis [13ŌĆō17] on the optimal operating conditions based on the interrelation between various parameters (e.g., flow rate and concentration of feed solutions, feed channel thickness, and pumping cost). Consequently, the gross power density has increased considerably since Pattle obtained power output of 0.2 W/m2 [3]. The best power output reported to date using the concentration difference by mixing 17 mM river water and 500 mM sea water is 2.4 W/m2 [11], which was achieved by using ultrathin pore-filling membranes and specific spacers with a high open area.
By comparison, the choice of redox species for RED in an electrode system is very limited. For instance, Fe2+/Fe3+ or ferri-/ferrocyanide [18ŌĆō21] are the most commonly used redox species in the aforementioned studies. These redox species have low overpotential for electron transfer, which leads to low electrode resistance and maximum electric power. Because the ferri-/ferrocyanide can be decomposed into cyanide under sunlight and oxygen, Fe2+/Fe3+ is regarded as the more stable redox species in pilot scale systems [19]. Recent papers [22,23] have reported large-scale RED systems utilizing Fe2+/Fe3+ redox couples with precious metal oxide electrodes under acidic pH media. However, several problems have been identified with such couples during long-term operation; for instance, iron precipitates can form when the pH is not properly controlled, leading to channel clogging, membrane fouling, and a decrease in current. In addition, leaking of the electrode solution into the discharge water is unavoidable due to the non-ideal permselectivity of ion exchange membranes [24,20,25].
Another redox reaction is water electrolysis, which generates hydrogen and oxygen at the cathode and anode, respectively. This reaction has been avoided due to its large overpotential and its products have thus far been regarded as waste gas. Further, hazardous gaseous mixtures of hydrogen and oxygen and toxic chlorine gas require proper collection/venting systems. However, water is a very safe chemical substance and does not give a negative impact on discharge water or the surrounding environment [26]. Moreover, hydrogen gas can be simultaneously obtained as surplus energy from water reduction at the cathode if the RED system is equipped with proper venting and collecting facilities. It is well known that hydrogen has been regarded as a promising energy carrier with good flexibility and the highest energy density on a mass basis [27].
A few studies on RED with water electrolysis have been reported to date (Table 1). Scialdone et al. demonstrated the performance of RED with oxidation/reduction of water and oxidation of chlorine in water/Na2SO4 or water/NaCl systems [26]. The maximum power density of 40 cells was roughly measured at 0.2 W/m2. Turek et al. simulated 5 cell pairs of RED in which the electrodes were connected to a rectifier to simulate an energy consumer, as the cell voltage did not otherwise exceed the overpotential for water electrolysis [28,29]. Some reports suggested special means to minimize the overpotential for water electrolysis, given the potential of the RED system as a method for renewable hydrogen gas production. For instance, the Logan group [30ŌĆō32] introduced bacteria on the anode to oxidize organic matter in a microbial RED that can avoid overpotential of the electrode reaction. Hatzell et al. utilized ammonium bicarbonate (AmB) [33,34] not only to create a salinity gradient but also to reduce the electrode overpotential for the hydrogen evolution reaction (HER). Chen et al.[35] reported that a conversion efficiency of hydrogen was close to 90% for 20 cells when a HCl solution was used as the catholyte, which minimized the overpotential for the HER.
However, these previous works were limited to small-scaled membrane stacks with fewer than 50 cells. In terms of a pilot-scale system, their purpose is contradictory to the general assumption [5,36,8,24] that electrode resistance caused by the overpotential of the electrochemical reaction can be minimized by using a large number of cell pairs (e.g., 50ŌĆō500). This means that minimizing the overpotential for the electrode reaction is not necessary for operating large-scale REDs. The report by Adriana et al.[25] on a pilot-scale RED with 500 cell pairs has addressed this point. When a NaCl solution was used as the electrolyte, the maximum power was 1.31 W/m2, which is only 13% smaller than that produced by the Fe2+/3+ redox system. They explained that the voltage loss by water electrolysis has a smaller effect on the overall cell voltage in the presence of a higher number of cell pairs. Practical cell voltages in industrial water electrolysis are ~1.8ŌĆō2.6 V [37], which are much smaller than the membrane voltage of RED with hundreds of cell pairs. We recently confirmed the difference in the maximum power between water electrolysis and ferri-/ferrocyanide is only 3% in a large-scale RED system with 1,000 cells when the flow rate of the electrode solution is set to be sufficiently low to maximize the net power [38]. This suggests that even though there is no specific strategy for minimizing the overpotential of water electrolysis, water oxidation/reduction occurs effectively at the electrodes in pilot-scale RED systems and can thus provide stable long-term operation without negatively affecting either the performance or the environment.
In this work, we present experimental results on the electric power and hydrogen production of a RED system feeding artificial NaCl solution with more than 50 cells in which the membrane voltage was large enough to achieve water electrolysis. Neutral-pH water containing small amounts of NaCl was used as the catholyte and anolyte, which is advantageous relative to strong acid or alkaline electrolytes as they are highly corrosive to the device, difficult to handle, and environmentally unfriendly [39]. The conditions employed herein are more universal, economical, and eco-friendly than previous studies (Table 1) on RED with hydrogen production. We confirm that hydrogen evolution occurs effectively at cathode due to its high stack voltage. To separate and collect hydrogen from oxygen or chlorine gas during RED operation, batch-type electrode chambers are used. The correlation of hydrogen energy with electric energy at maximum power is evaluated and compared with a conventional RED system circulating electrolyte to discern the effect of electrode resistance on the electric power relative to the number of cell pairs. Broadly, this study is aimed at the long-term operation of pilot-scale REDs with water electrolysis.
2. Experimental
2.1 Reverse electrodialysis (RED) operation
The membrane stack of the RED system consisted of cation exchange membranes (CEM-Type 1, Fujifilm, Netherlands), anion exchange membranes (AEM-Type 1, Fujifilm), spacers (thickness 100 ╬╝m), and gaskets (PTFE, thickness 100 ╬╝m). Artificial sea water (0.5 M) and river water (17 mM) were used by dissolving NaCl salt (Daejung, Republic of Korea) in the waters. River and sea waters enter their respective compartments using peristaltic pumps (Masterflex, Cole-Parmer, US) at a flow rate of 4 mL minŌłÆ1 per cell. Distilled water with a pH of ~6 as the electrode solution was added in batch-type chambers (50 mm ├Ś 50 mm ├Ś 50 mm) (Fig. 1b, termed batch-type RED). Gas bags (Dalian delin gas packing co., ltd, China) were connected to the chambers to collect the produced gas (See a photo of the real RED system in supporting information, Fig. S1). For comparison, the performance of a conventional RED (Fig. 1c) was also evaluated, in which thin (1 mm) electrode chambers were used. The electrode solution was circulated from the anode to the cathode at a flow rate of 50 mL minŌłÆ1 and gas collection was achieved using a container connected to the electrode chambers. Platinum-coated titanium mesh (thickness 1 mm, geometric area 19.625 cm2) was used as the anode and cathode in both types of RED systems. The effective area of an individual membrane in the batch-type RED was 19.625 cm2 (3.14 ├Ś 2.5 cm ├Ś 2.5 cm), which is identical to that of the conventional RED system.
2.2 Performance evaluation
A potentiostat (ZIVE SP2, Wonatech, Republic of Korea) was used to measure polarization (IŌĆōV) curves with varied cell pairs at a scan rate of 40 mV sŌłÆ1. The maximum power (Pmax) and the corresponding current density were determined from peak values of the parabolic curves. The measured Pmax was converted to the maximum power density (Pd,max) based on the cell pair area.
Where Nm is the number of cell pairs and A is the effective area of individual membrane.
Applying a constant load at which maximum power was achieved in the IŌĆōV curves, we evaluated the average electric energy and the amount of hydrogen of the two types of RED systems with different cell pairs (25, 50, 75, and 100). All measurements were performed in duplicate with the low variability (~5%). Ion chromatography (ICS-1600, Thermo scientific, USA) was used to analyze the ion concentrations of the catholyte and anolyte after 1 h of the constant load tests.
2.3 Hydrogen production rate and hydrogen energy
The produced gas was collected in the gas bag of the electrode chamber during the constant load test for 1 hour. And then the gas bag was connected to a gas chromatography (GC 2014, Shimadzu, Japan) to evaluate the gas component and the amount of hydrogen. Hydrogen production rate (mL hŌłÆ1 cmŌłÆ2-electrode) was calculated as the measured hydrogen volume was divided by the geometric area of electrode.
The energy extracted from the produced hydrogen was calculated based on the volume-specific chemical energy at standard temperature and pressure (STP) conditions [40]. Given that the product water in fuel cells is produced as a liquid (2), we used the higher heating value (HHV, 285.83 kJ/mol) of hydrogen to calculate its chemical energy:
in which the molar gas volume at STP is V0,mol = 22.414 Nl/mol and Nm3 is normal cubic meters.
The current efficiency was calculated as the experimental hydrogen production rate was divided by the theoretical rate (mL hŌłÆ1 cmŌłÆ2-electrode).
Where I is the measured current, n is the stoichio-metric number of electrons consumed in the electrode reaction (n = 2 for hydrogen production from water electrolysis), F is the Faraday constant, A is the geometric area of electrode.
2.4 Theory: voltage and resistance
The membrane voltage with no current, open circuit voltage (OCV), produced by one cell consisting of an AEM and a CEM is given by the Nernst equation [41].
where z is the ion valence (i.e., 1 for NaCl), F is the Faraday constant, R is the universal gas constant, and the factor of 2 accounts for the CEM and AEM. ╬▒ is the permselectivity of the ion exchange membrane and a is the activity of the concentrated (ac) and diluted (ad) solutions.
The membrane voltage of the stack consisting of the total number (Nm) of cell pairs is:
The current through an ideal RED is given by [42]:
where Ri is the internal resistance of the stack and Ru is the external load resistance.
The produced electric power (Pu) from Ru is:
Pu approaches its maximum value when Ru is equal to Ri. In this case the Estack,Pmax is half the value of Em,OCV, where Estack,Pmax is the voltage between the cathode and anode at maximum power.
Ri can be divided into ohmic area resistance (Rohmic) and non-ohmic resistance (Rnon-ohmic). Rnon-ohmic considers the concentration change in the boundary layers (RBL) and in the bulk solution (RΔC). Rohmic is determined by the resistances of the membranes (RAEM and RCEM), two feed solutions (Rriver and Rsea), the electrode system (Relectrode), and the number (Nm) of cell pairs [43]:
The voltage loss (Relectrode ┬Ę I) of the electrode system originates from the thermodynamic electrode potential, overpotential, and concentration polarization for the redox reaction. We measured the voltage loss by using Ag/AgCl reference electrodes (Fig. S2), which read the membrane voltages of the stack at maximum power (Em, Pmax):
2.5 Gross energy efficiency
The maximal available free energy per second (Xmax) was calculated from the Gibbs free energy of mixing river water with sea water [2].
where R is the universal gas constant (8.314 J molŌłÆ1 KŌłÆ1) and T is the temperature (K). CR and CS are the NaCl concentrations (mol mŌłÆ3) of river water and sea water, respectively, and ╬”R and ╬”S are their respective flow rates (m3 sŌłÆ1). CM is the equilibrium concentration (CM = (╬”RCR + ╬”SCS)/(╬”R + ╬”S)).
The gross energy efficiency is defined as follows:
3. Results and Discussion
Fig. 1a shows the flow of counter-ions across the ion exchange membranes by the concentration difference in the RED system. When distilled water is used as the electrolyte, the electrode chamber of batch-type RED (Fig. 1b) does not ideally contain chloride ions during operation because a CEM was used as the shielding membrane. In actual experiments, however, a considerable amount of NaCl salt was found in the catholyte and anolyte during RED operation. Ion chromatography (IC) showed that the cathode and anode chambers adjacent to the sea and river waters contained ~40 mM and ~2.5 mM NaCl, respectively, during 1 h of operation. There are two possible reasons for this observation; the first concerns the non-ideal property of ion exchange membranes; hence, co-ion transport by imperfect membrane selectivity must be considered. Another reason for the observation of NaCl is leakage of the sea and river waters into the electrode chambers through small gaps between the gaskets and spacers due to pressure imbalance over the membrane area.
Given the inflow of NaCl into the electrode chamber, when water electrolysis is used as the redox reaction for RED operation, the cathodic and anodic reactions in neutral pH condition are represented as follows [44]:
Cathode:
Anode:
The reduction reactions of water (13) and oxygen (14) occur at the cathode and NaCl does not participate in electrochemical reduction. At the anode, however, water oxidation (15) competes with the oxidation of chloride anions (16).
In conventional RED with electrolyte circulation (Fig. 1c), the current and electric power are influenced by the number of protons (17), hydroxide (18), and hydrogen (19) dispersed in solution, which are products of water reduction/oxidation. They move in the opposite electrode chamber by convection and therefore undergo electron transfer reactions at the anode and cathode as:
Cathode:
Anode:
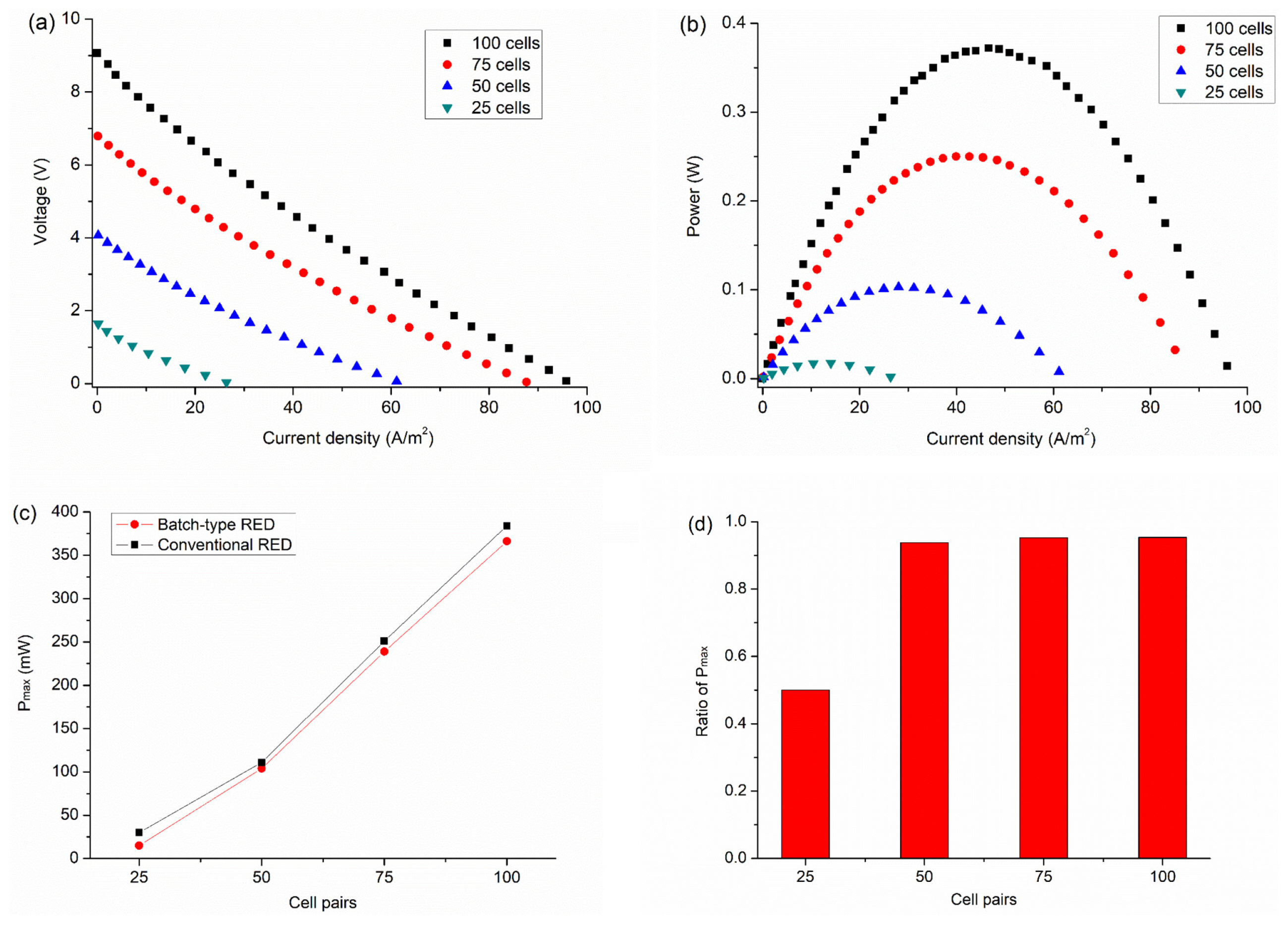
The polarization curves (Fig. 2a) were measured to evaluate the effect of the number of cell pairs on the power output of RED system using water electrolysis as the electrode reaction. The maximum current density (jmax) increased from 26 A/m2 to 95 A/m2 with the number of cell pairs. On the other hand, an increment in jmax decreased with increasing cell pairs, which can be ascribed to the leakage current that passes through the feed and drain channels. Previous research showed that the leakage current increases with the increase of the number of cell pairs and causes the current efficiency to decrease [42,45]. Fig. 2b shows the measured power as a function of the current density derived from Figure 2a. The Pmax increased from 15 mW to 366 mW as the number of cell pairs increased from 25 to 100. The current density (Fig. S3) and Estack (Fig. S4a) at Pmax also increased with increasing cell pairs. This indicates that increasing the stack voltage activates water decomposition, leading to increased electric power. We also monitored Em,OCV and Em(Fig. S4a) at Pmax by using two Ag/AgCl reference electrodes during the IŌĆōV curve measurements. The difference between Em and Estack at Pmax indicates the voltage loss (Relectrode ┬ĘI ) by water electrolysis, which was ~2.2 V regardless of the number of cell pairs. Compared with Em at Pmax with 25 cell pairs, the voltage loss of water electrolysis is relatively large, leading to a voltage ratio of 0.80 (Fig. S4b). As the number of cell pairs increases, however, the contribution of voltage loss by water electrolysis diminishes, leading to a decrease in the voltage ratio to a value of 0.35.
Fig. 2c shows plots of maximum power (Pmax) with respect to the number of cell pairs of batch-type and conventional RED systems. The Pmax of conventional RED also increased proportionally with the number of cell pairs. Fig. 2d shows the Pmax ratio of batch-type RED to conventional RED derived from Fig. 2c. Compared with conventional RED with 25 cells, the electric power of the batch-type RED system was decreased by half, which we attribute to a decrease in electrode resistance by electrolyte circulation. The reaction products (i.e., H+ ions) at the anode in conventional RED move into the cathode chamber and become electroreduced at the cathode, producing hydrogen. Because the overpotential for H+ reduction is much lower than that for water reduction, the current and electric power increase. An analogous process occurs for the electrooxidation of OHŌłÆ ions at the anode. However, the effect of electrolyte circulation was markedly decreased when more than 50 cell pairs were employed. The difference in electric power between the batch-type and conventional RED systems was only 5%. This means that the Em,Pmax (Fig. S4a) for more than 50 cells is sufficient to achieve electrochemical oxidation and reduction of water molecules in neutral-pH media.
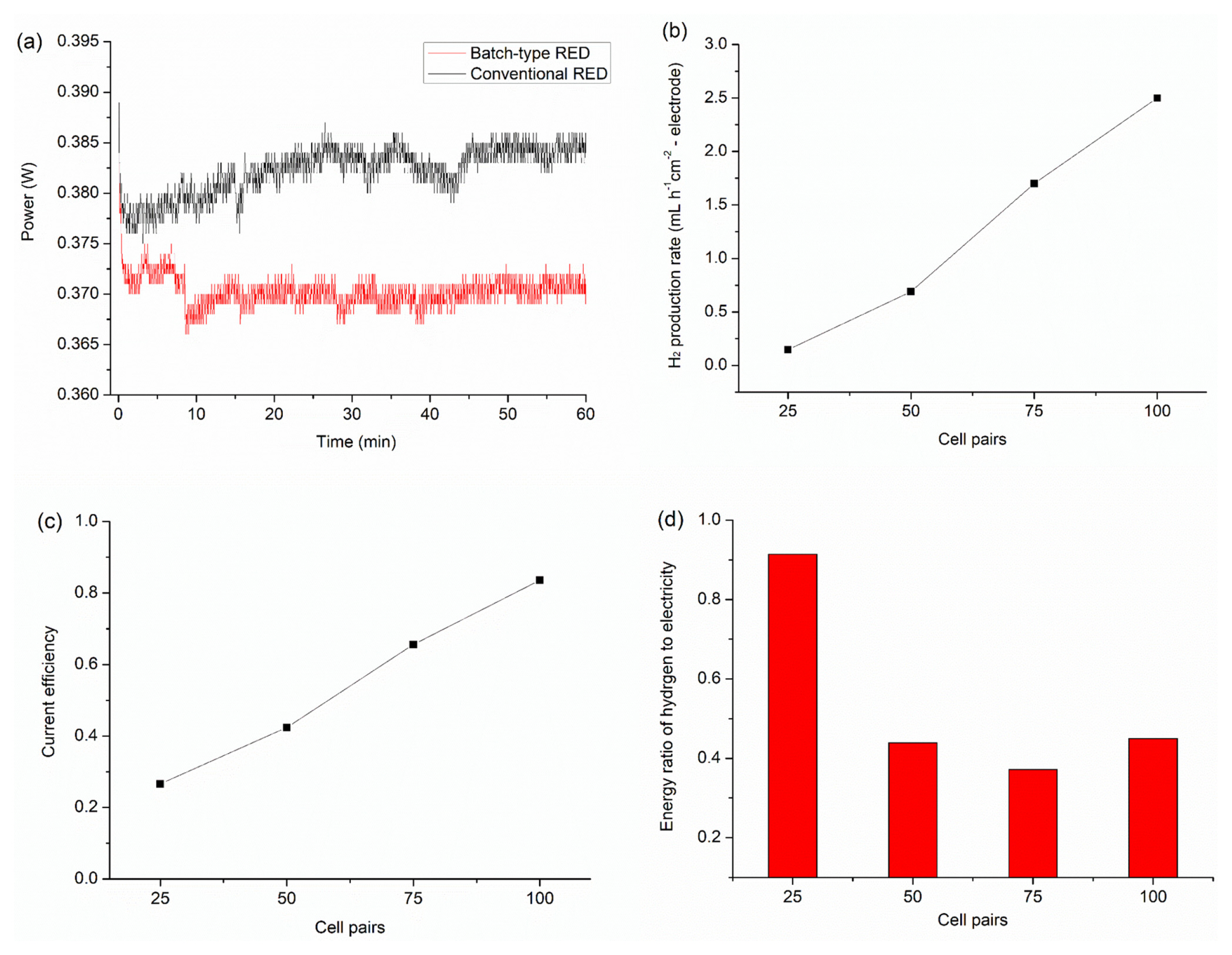
To monitor average electric energy and hydrogen production rate of batch-type RED, the constant external load at which maximum power was obtained in the IŌĆōV curves was applied for 1 h (Fig. 3a). Gases produced at the cathode and anode were collected separately. The RED with 100 cells had the largest concentration percentage of hydrogen (71%) (Table S1). The remaining gases were oxygen and nitrogen. As mentioned above, leakage of sea and river waters into the electrode chamber occurred during RED operation. The water level within the chamber slowly rose, which caused the air in the cylindrical connector to diffuse into the gas pack during the constant load experiments. This phenomenon can explain why the concentration percentage of hydrogen was less than 100%. If the leakage problem were solved, the production of highly pure hydrogen would be expected in the batch-type RED. From the concentration percentage, the hydrogen production rates with different cell pairs were calculated (Fig. 3b). Overall, the production rate was proportional to the number of cell pairs and current density. A hydrogen production rate of 2.5 mL cmŌłÆ2 hŌłÆ1 was obtained for 100 cells, corresponding to 1.1 ├Ś 10ŌłÆ4 mol cmŌłÆ2 hŌłÆ1, which is larger than previously reported values for RED-driven hydrogen production [46,33,6,34]. Tufa et al.ŌĆÖs work [47] showing high hydrogen production rate (2.0 ├Ś 10ŌłÆ3 mol cmŌłÆ2 hŌłÆ1) is out of scope for our study. Because hydrogen was produced from alkaline polymer electrolyte water electrolysis operated by excess power of RED, which is not simultaneous production of hydrogen during RED operation. It should be noted that this high hydrogen production rate was achieved from water electrolysis in NaCl solution at maximum power of RED with 100 cell pairs feeding an artificial NaCl solution with a salinity ratio of 30. Compared with previous research (Table 1), our system is more universal, economical, and eco-friendly, which are requisite conditions for the development of scaled-up RED systems with hundreds of membrane pairs.
The current efficiency, which is the ratio between the experimental hydrogen production rate and theoretical one, was calculated for batch-type RED systems with varied cell pairs (Fig. 3c). As the cell pairs increased, the current efficiency steadily increased. Notably, the electric power decreased by half when N2 purged aqueous solution was used, indicating that some of the parasitic current originated from the oxygen reduction reaction (ORR). However, the effect of this parasitic current decreased as the number of cell pairs increased, with the current efficiency approaching 0.85 for the RED system with 100 cells. This result demonstrates that water reduction predominantly occurred at the cathode under large stack voltage. The batch-type RED with more than 120 cells is expected to show near 100% conversion efficiency. We confirmed that gas crossover caused by leakage between the cathode and anode chambers was negligible on the basis of the concentration percentage of hydrogen in the gas pack of the anode chamber being slightly larger than that of the air.
Fig. 3d shows the hydrogen-to-electric energy ratios of the batch-type RED. The ratio for 25 cells was ~0.9, which suggests that a considerable amount of Em,Pmax was applied at the electrode due to high electrode resistance, which arises from the relatively large overpotential of water electroreduction/oxidation. At a result, the voltage (Estack,Pmax) applied at the external resistance was much smaller than Em,Pmax (Fig. S4a), indicating that water electrolysis disturbed production of electric power. At more than 50 cell pairs, however, the ratio of hydrogen energy remained ~0.4. As the number of cell pairs increased, the contribution of the electrode resistance to the total resistance (including AEM, CEM, sea water, and river water) became much smaller. Moreover, Em,OCV of more than 50 cell pairs is large enough to achieve water electrolysis. Therefore, water electrolysis is an available redox reaction to generate electric power in RED with a sufficient number of cell pairs. In addition, this condition decreased the susceptibility of water electrolysis (or hydrogen production) to the pH of the electrode solution. When starting the constant load test, electrode solution was distilled water with a pH of ~ 6. As the RED system operates, the catholyte and the anolyte become more basic and acidic, respectively. The resultant pH values of the catholyte and the anolyte were 12 and 3, respectively. Nevertheless, 100 cells of batch-type RED maintained a constant current for 1 h (Fig. 3a). This indicates that the electrochemical reduction/oxidation of water molecules occurred successfully in the presence of the large RED stack voltage even though the pH of the electrolyte changes sharply. This abrupt change in pH when using the batch-type electrode chamber can be mitigated by switching feed solutions on a regular cycle, which leads to electric current in the opposite direction.
We also conducted constant load experiments of conventional RED (Fig. 3a, black line) with different cell pairs for 1 h. Fig. 4 shows the hydrogen energy of batch-type and conventional RED systems with varied cell pairs. As seen, the hydrogen energy of batch-type RED was larger than that of conventional RED for all cell pairs, and the difference in the hydrogen energy became larger as the number of cell pairs increased. It is expected that as more cell pairs are added to a conventional RED, more hydrogen will be dispersed into solution and circulate between the anode and cathode; it will then oxidize to produce protons at the anode, where the overpotential is much lower than water oxidation. This is why the electric energy of conventional RED is slightly larger than batch-type RED and the amount of hydrogen in the former was smaller than that in the latter.
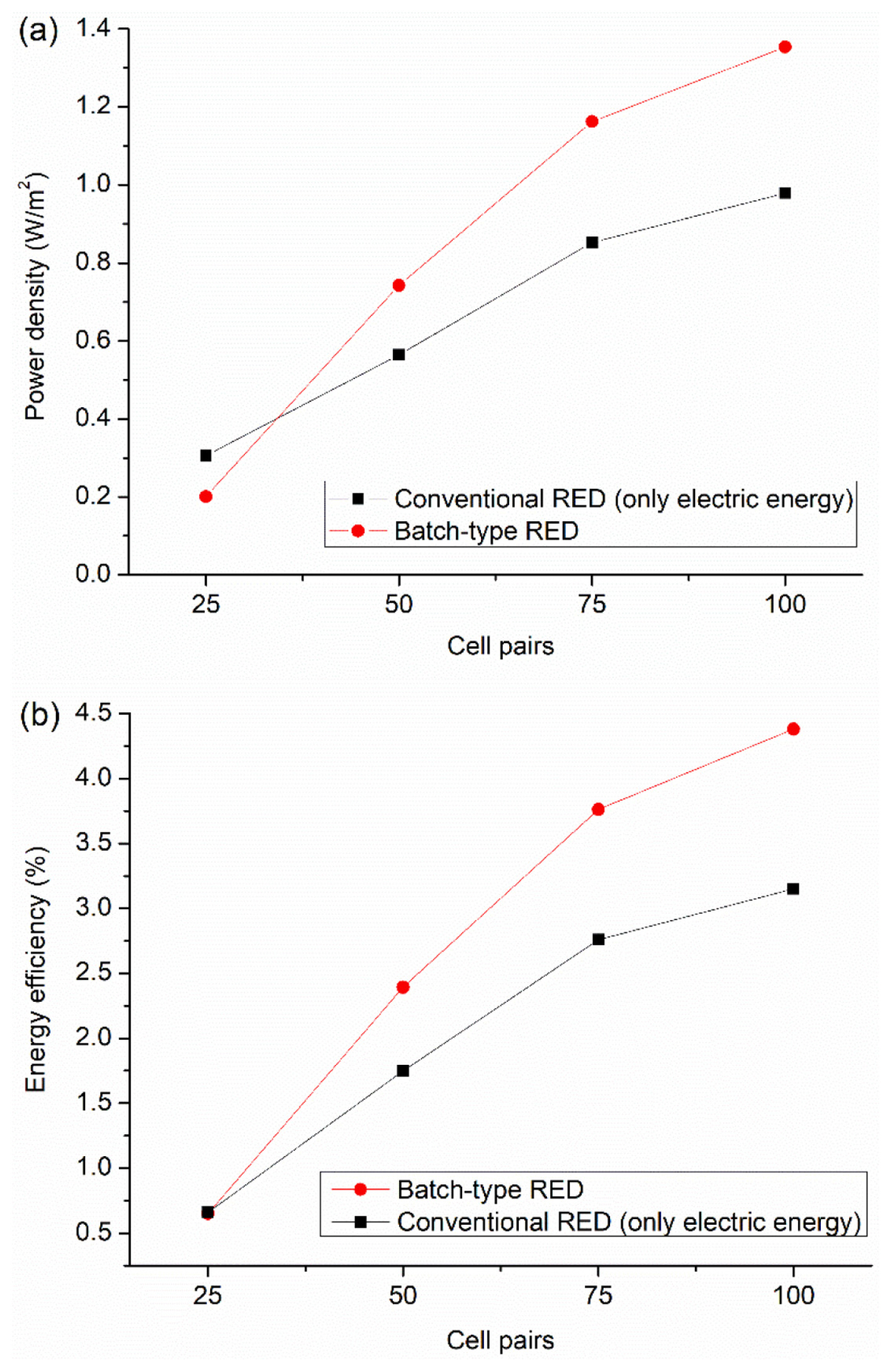
It should be noted that while hydrogen collected from batch-type RED is expected to be pure, the gas pack of conventional RED contains a mixture of hydrogen and oxygen gas. Thus, hydrogen produced from conventional RED should be separated from oxygen and other gases, which means conventional RED requires installation and maintenance costs for the gas separation device. Moreover, the hydrogen and oxygen mixture is potentially explosive when its minimum activation energy is reached. Therefore, it could be more economical and safer to produce only electric power in conventional RED. Fig. 5a shows a comparison of power density between batch-type and conventional RED systems, with only electric energy considered for the latter. As seen, the increase in power density of batch-type RED was larger than conventional RED as the number of cell pairs increased. At 25 cells, where the practical voltage for water oxidation/reduction is relatively large compared to membrane voltage, the power density of batch-type RED was lower than conventional RED. However, RED with more than 50 cells had sufficient driving force to achieve water electrolysis; all hydrogen gas produced at the cathode can be available as energy, leading to an increase in the power density, unlike conventional RED with electrolyte circulation where a considerable amount of hydrogen is consumed at the anode. This trend was directly reflected in the gross energy efficiency (Fig. 5b). As the number of cell pairs increased, (1) the energy efficiency of both systems sharply increased, indicating that a larger stack voltage reduces the effect of electrode resistance on the electric power, (2) the difference in energy efficiency between the batch-type and conventional systems became larger, which means the contribution of hydrogen energy to the total energy was larger.
4. Conclusions
We have demonstrated that even though there is no specific method to reduce overpotential for water electrolysis, high membrane voltage of RED with more than 50 cells can utilize the water oxidation/reduction in neutral-pH media containing small amounts of NaCl as electrode reaction. As the number of cell pairs increased, the effect of voltage loss by electrode resistance on the produced electric power became negligible. Therefore, for stable long-term operation of the pilot-scale RED systems consisting of hundreds of cell pairs, water electrolysis with a neutral-pH electrolyte (e.g. tap water) can be an available redox reaction to produce not only electric power but also hydrogen energy. Moreover, non-noble metal catalysts (e.g., Ni, Mn, and Ti) [48,35] can be available as cathodes for water electrolysis because the high membrane voltage can compensate for low electrocatalytic ability of these metal catalysts. We recently confirmed that Ti mesh can be used for a cathode for 1,000 cell pairs of RED without loss of power [38]. If the properties of the ion exchange membrane and channel design are optimized to increase the current density of the RED system, it is expected that pure hydrogen can be produced at a higher production rate from RED systems. We expect that pilot-scale RED operation with simultaneous hydrogen production will be a representative example of the successful combination [37,49ŌĆō51] of water electrolysis and renewable energy.