Synthesis of Novel (Be,Mg,Ca,Sr,Zn,Ni)3O4 High Entropy Oxide with Characterization of Structural and Functional Properties and Electrochemical Applications
Article information
Abstract
The new emerging “High entropy materials” attract the attention of the scientific society because of their simpler structure and spectacular applications in many fields. A novel nanocrystalline high entropy (Be,Mg,Ca,Sr,Zn,Ni)3O4 oxide has been successfully synthesized through mechanochemical treatment followed by sintering and air quenching. The present research work focuses on the possibility of single-phase formation in the aforementioned high entropy oxide despite the great difference in the atomic sizes of reactant alkaline earth and 3d transition metal oxides. Structural properties of (Be,Mg,Ca,Sr,Zn,Ni)3O4 high entropy oxide were explored by confirmation of its single-phase Fd-3m spinel structure by x-ray diffraction (XRD). Further, nanocrystalline nature and morphology were analyzed by scanning electron microscopy (SEM). Among thermal properties, thermogravimetric analysis (TGA) revealed that the (Be,Mg,Ca,Sr,Zn,Ni)3O4 high entropy oxide is thermally stable up to a temperature of 1200°C. Whereas phase evolution in (Be,Mg,Ca,Sr,Zn,Ni)3O4 high entropy oxide before and after sintering was analyzed through differential scanning calorimetry (DSC). Electrochemical studies of (Be,Mg,Ca,Sr,Zn,Ni)3O4 high entropy oxide consists of a comparison of thermodynamic and kinetic parameters of water and hydrazine hydrate oxidation. Values of activation energy for water oxidation (9.31 kJ mol−1) and hydrazine hydrate oxidation (13.93 kJ mol−1) reveal that (Be,Mg,Ca,Sr,Zn,Ni)3O4 high entropy oxide is catalytically more active towards water oxidation as compared to that of hydrazine hydrate oxidation. Electrochemical impedance spectroscopy is also performed to get insight into the kinetics of both types of reactions.
1. Introduction
To meet the demands of growing technology, material scientists are in a continuous struggle to develop new materials that overcome the impediments offered by conventional materials. High entropy materials [HEMs] is a new concept in the scientific world. In contrast to conventional materials, HEMs consist of (a) five or more major reactant components with at. % ranging from 5 to 35% and (b) possess configurational entropy greater than 1.5 R without showing any dependence on phase [1,2]. Among HEMs, high entropy alloys [HEAs] were synthesized for the very first time in 2004 [3,4]. Later on, the idea of HEAs was extended to (a) high entropy ceramic materials [HECMs] like high entropy oxides [HEOx], high entropy borides [HEBs], high entropy carbides [HECs] (b) high entropy polymers [HEPs] and (c) high entropy composites [HECOMPs]. From this, the new field of HEMs was emerged [5].
Entropy is the main driving force that is responsible for the formation of solid solutions and simpler crystal structures such as face-centered cubic (FCC) or body-centered cubic (BCC) [6]. High entropy of mixing and a definite temperature causes equiatomic multicomponent alloys to form a solid solution rather than forming a mixture of intermetallic compounds [7]. These materials exhibit marvelous corrosion-resistant properties [8] along with exceptional thermal stability [9] and structural strength, etc [10,11].
Among HEOx, Rost et al successfully synthesized single-phase high entropy metal oxide (MgNiCo-CuZn)O with FCC structure for the first time in 2015 [12]. Sicon Jiang introduced entropy stabilized single-phase perovskites i.e., Sr1(ZrSnTi-HfM)1O3, Ba1(ZrSnTi-HfM)1O3 [M = 0, Mn, Ce, Y, Ge, Nb] & (SrBa)1(ZrSnTi-HfNb)1O3 having complex ionic structure with at least two cation sublattices [13]. Further extension to this field includes the successful fabrication of the following HEOx: (a) A(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 [A = Gd, La, Nd, Sm, Y] [14] (b) (Sc0.2Ce0.2Pr0.2Gd0.2Ho0.2)2O3 [15] (c) (CeGdLaNdPrSmY)O [16] (d) (MgCoNiCuZn)1−x LixO, and (MgCoNiCu)0.8(LiGa)0.2O [17].
From the applications point of view, HEOx is reported to possess larger colossal dielectric constant [17], superior ionic mobility [18], high energy storage capacity and cycling stability [19] as compared to that of conventional oxides. Therefore, the potential of HEOx can effectively be exploited by using them in capacitors and reversible energy storage devices. Irrespective of all these achievements, the research on HEMs is restricted to one or two fields and most of it needs to be revealed. This wide hole compels the material scientists to expose the other aspects of HEMs so that the bottlenecks encountered by the conventional materials can be overcome. Keeping this in view, we explored the tendency of single-phase formation in a hexanery equiatomic (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx synthesized by a beautiful blend of alkaline earth and 3d transition metal reactant oxides for the first time. Thermal properties reveal trends of its thermal stability across the whole temperature range under study. Moreover, the electrochemical studies unveil the efficiency of the novel (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx as an electrocatalyst for water and hydrazine hydrate oxidation. A comparison of these results shows its potential in electro-oxidation of these two species.
2. Materials and Methods
2.1 Synthesis
Present research work is focused on the synthesis of a novel equiatomic HEOx through solid-state reaction. Magnesium oxide (MgO), calcium oxide (CaO), strontium carbonate (SrCO3), nickel oxide (NiO), beryllium oxide (BeO), and zinc oxide (ZnO) were taken as precursor materials and mixed in equiatomic ratio. These metal oxides were mixed homogeneously for 6 hours at 250 rpm in a planetary ball mill. The milling was interrupted for 3 minutes after every 12 minutes milling to avoid a sidestep rise in the vial temperature. Silicon carbide balls were used as milling media. The milled powders were cold compacted into 0.5 gram pellets by applying a pressure of 4 kN. These green compacts were sintered at 1150°C at a heating rate of 10°C/min for 10 hours in a chamber furnace followed by air quenching to lock the crystal structure.
2.2 Instrumentation
Pan analytical x ray diffractogram (XRD) equipped with Cu-Kα radiation (λ= 1.540598) was used to study the crystalline nature of the synthesized sample having a step size of 0.02°. Morphology of the sample was analyzed through MIRA 3 TESCAN Field Emission Scanning Electron Microscope (SEM). Thermal stability of the material before and after sintering was studied by Thermal gravimetric analysis (TGA) and Differential Scanning Calorimetry (DSC) that was carried out on STARe SW12.10. The TGA and DSC analyses were performed in an open-air atmosphere at a heating rate of 10° min−1 from room temperature to 1200°C and 600°C respectively.
Electrochemical studies i.e. water oxidation and hydrazine hydrate oxidation were performed on Gamray Interface 1000. The measurements were carried out in a three-electrode cell consisting of modified glassy carbon (GC) as a working electrode, gold as a counter electrode, and silver-silver chloride (Ag/AgCl) as a reference electrode. Hydrazine hydrate oxidation was carried out in 0.1 M KOH solution in a potential window ranging from −1.0 V to 0.8 V at different scan rates. While the concentration effect was investigated at a scan rate of 10 mV s−1. For electrochemical impedance, DC and AC voltage was kept at 0 V and 800 V respectively at a frequency of 0.1 Hz to 1 M Hz.
For water oxidation, the silver wire was used as the counter electrode while keeping the working and reference electrodes constant as used for hydrazine hydrate oxidation. The scan rate effect was studied in 0.1 M KOH solution while varying the potential window from 0 V to 1.7 V. While an AC voltage of 1.7 V is found appropriate for carrying out the impedance studies in a frequency range of 0.1 Hz to 1 M Hz.
2.3 Modification of the electrode
Before electrode modification, it should be ensured that the surface of the GC is free from any deposition. In the case of any adsorption, the electrode should be immersed in a solution of sulfuric acid and take cyclic voltammograms in the full window at 100 mV s−1. GC is modified by placing finely ground 5 mg of the catalyst on the working surface of the electrode. This is followed by the drop-casting of 10 μL of slurry containing 0.5% Nafion in methanol as shown in Fig. 1 [20]. The electrode surface is dried by placing it in an oven at 55°C for 1 hour. Preconditioning is done at 100 mV s−1 in the working potential window to activate the catalyst surface and try to get stable voltammograms.

A series of different steps involved in the modification of the electrode.
3. Results and Discussion
The schematic diagram as shown in Fig. 2 presents the pattern of the present study. The different sections of the study are shown below:

Schematic illustration of the structural properties, thermal properties and electrochemical applications of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx.
3.1 Structural Properties
3.1.1 Crystal Structure Analysis
Fig. 3 (a) shows that the powder mixture before milling is mere an unreacted amalgam of metal oxides exhibiting characteristics peaks of reactant metal oxides. XRD patterns in Fig. 3 (b) denote the course of formation of a single-phase when the sample is milled and sintered at different temperatures followed by air quenching. Subjecting the powder mixture of reactant metal oxides to mechanochemical treatment in a planetary ball mill for 6 hours compels them to lose their identities and undergo structural evolution towards the formation of a single-phase. However, when the sample is sintered and air quenched at 1050°C, we experience the presence of more than one phase in the crystal structure. But with the rise in sintering temperature up to 1150°C, no secondary phases appear and only a single-phase is formed with a clear Fd-3m spinel structure. Although all the considered metal cations exhibit +2 oxidation state yet they form spinel Fd-3m structure rather than rock salt Fm-3m. Generally, metal ions with +2 valency may crystallize into rock salt structure in metal oxides. In ceramics, the partial pressure of oxygen plays an important role and may induce the tendency for the formation of mixed spinel phases. But the XRD results in Fig. 3 (b) clearly show that a single-phase Fd-3m spinel structure is formed with (Be,Mg,Ca,Sr,Zn,Ni)3O4 stoichiometry [21]. The lattice parameter of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx comes out to be 9.19 A° along 310 plane.
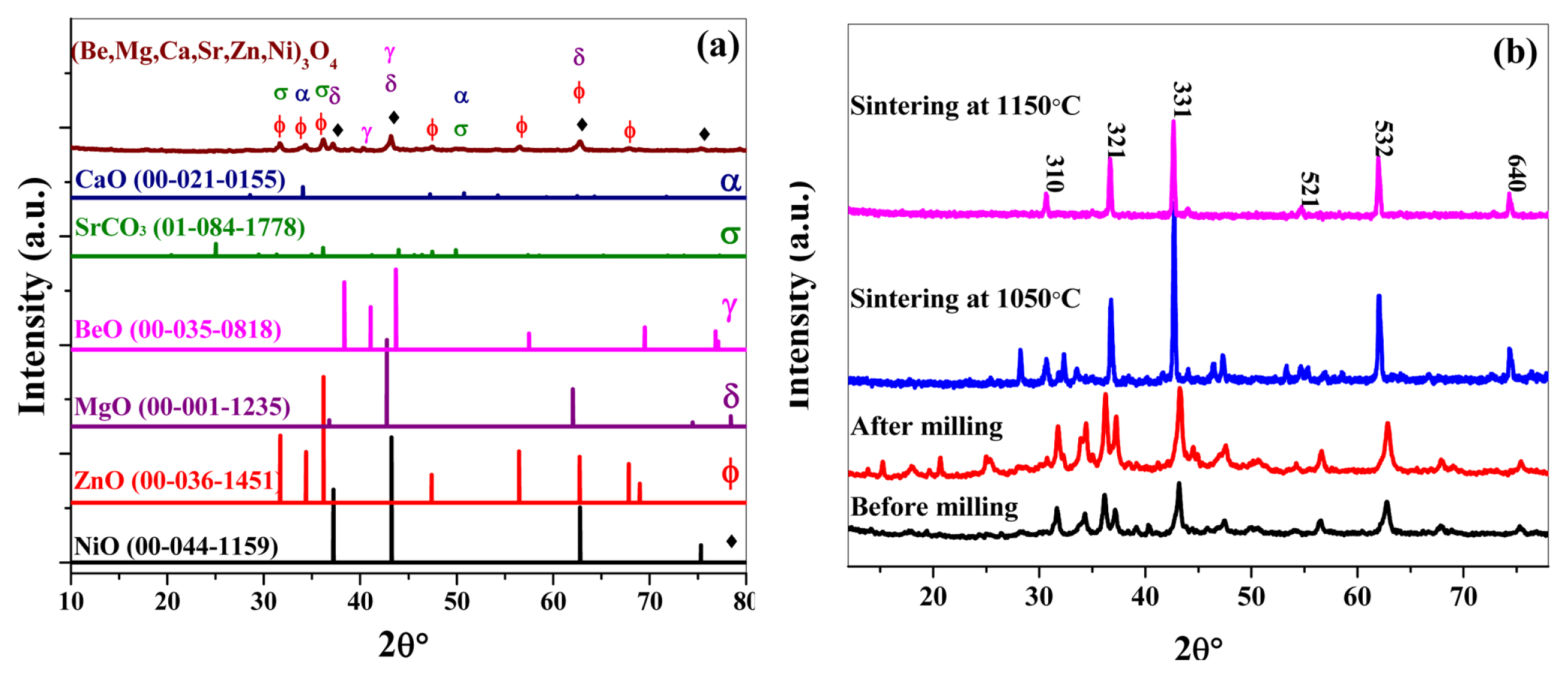
(a) Comparison of the XRD pattern of a precursor powder mixture of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx with the standard XRD spectra of all the precursor metal oxides before milling (b) Role of milling and temperature conditions in the formation of single-phase HEOx.
Table 1 shows that the milling process leads to a decrease in the particle size as well as an escalation in the lattice strain. Usually, a decrease in particle size and increase in lattice strain is observed after sintering. But, a further reduction in the particle size and lattice strain is observed when the sample is sintered at 1150°C followed by quenching in air. This exceptional behavior is justified because larger-sized particles possess larger quench-induced stress and are easily cracked [22].
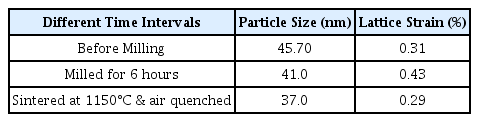
Effect of ball milling and sintering conditions on the particle size and lattice strain of (Be,Mg,Ca,Sr, Zn,Ni)3O4 HEOx.
Particle size plays a key role in determining the efficiency of a catalytic process whether it is heterogeneous or homogeneous catalysis. Heterogeneous catalysis is carried out at the solid/liquid or liquid/gas interface and is greatly influenced by the crystallite size of the catalyst. Size-dependent studies of catalysts showed that catalytic activity is the function of size, coordination number, and shape of the crystallite. When the catalyst size approaches in the nanometer regime, the co-ordination number decreases, and the effect of atoms at the corners and edges become dominant. Atoms having low co-ordination numbers adsorb strongly to the reactant molecules, decrease their activation energy, and accelerate the catalytic reaction [23]. The catalytic response of IrO2 per gram of catalyst is significantly improved by decreasing its particle size to 1.6 nm [24]. Keeping in view these studies, it can be anticipated that the catalytic activity of the (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx increases with the decrease in the particle size. Table 1 shows that the blend of precursor (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx material has maximum crystallite size. Six hours of mechanical milling leads to a reduction in the particle size and a maximum decrease in size is observed after sintering. Hence, the HEOx with the smallest crystallite size is employed as the catalyst for water and hydrazine hydrate electrochemical studies to attain the best catalytic activity.
3.1.2 Thermodynamic Analysis
In HEMs, configurational entropy is the key factor responsible for the elimination of most of the secondary phases and the formation of a single-phase in resultant compounds. Thermodynamically, two opposite terms compete for each other to define the resultant phase in a multi-component system [25]: (a) enthalpy of mixing (ΔHmix) and (b) entropy of mixing or configurational entropy (ΔSmix). ΔHmix usually hinders the formation of a solid solution while ΔSmix favors the solid solution formation. ΔSmix of a system can be calculated by using the following equation [19]:
Where, xi and xj are the mole fractions of ions present at the cation and anion site respectively. The entropy of mixing for (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx system comes out to be “5.8 R”. This is an appreciable amount to ensure the negative value of Gibbs free energy (ΔGmix) of reaction at high temperatures. Hence, we can expect the formation of a single-phase solid solution in (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx system. However, the sintering temperature for the formation of single-phase needs to be optimized as we can see in the XRD patterns of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx system in Fig. 3 (b). In the HEOx system under study, 1150°C comes out to be an optimum temperature for the formation of a single phase.
3.1.3 Microstructure Analysis by SEM
SEM micrographs in Fig. 4 reveal that apparently (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx consists of microparticles of irregular shape. But on further magnification, it becomes clear that these microparticles are in fact agglomerates of nanoparticles. Typically microparticles are formed when the solid-state method is adopted as a mode of synthesis. But as we subjected the sintered sample to air quenching, the particle size is reduced up to the nanometer scale as explained by both XRD and SEM results. From the nanocrystalline nature of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx, its potential catalytic activity can be anticipated due to the larger surface area.

SEM images of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx at different magnifications.
3.2 Thermal Properties
3.2.1 Thermogravimetric Analysis
Thermogravimetric analysis was performed to study the variation in the thermal behavior of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx before and after sintering. Before the thermal treatment of the sample, three weight losses were observed as depicted in Fig. 5 (a). The first weight loss observed was 16% ranging from room temperature to 120°C. This corresponds to the desorption and evaporation of the species that are physically bound to the surface of the oxide. Further, 2% weight loss was observed in the range of 361°C to 392°C and is attributed to the removal of chemically bound water molecules. Both of these weight losses are complemented by two endothermal peaks in the DSC curve as shown in Fig. 5 (b). Then, a steep weight loss of 6% in the range of 783°C to 879°C is ascribed to the decomposition of the reactant strontium carbonate [26,27] and the formation of the solid solution.
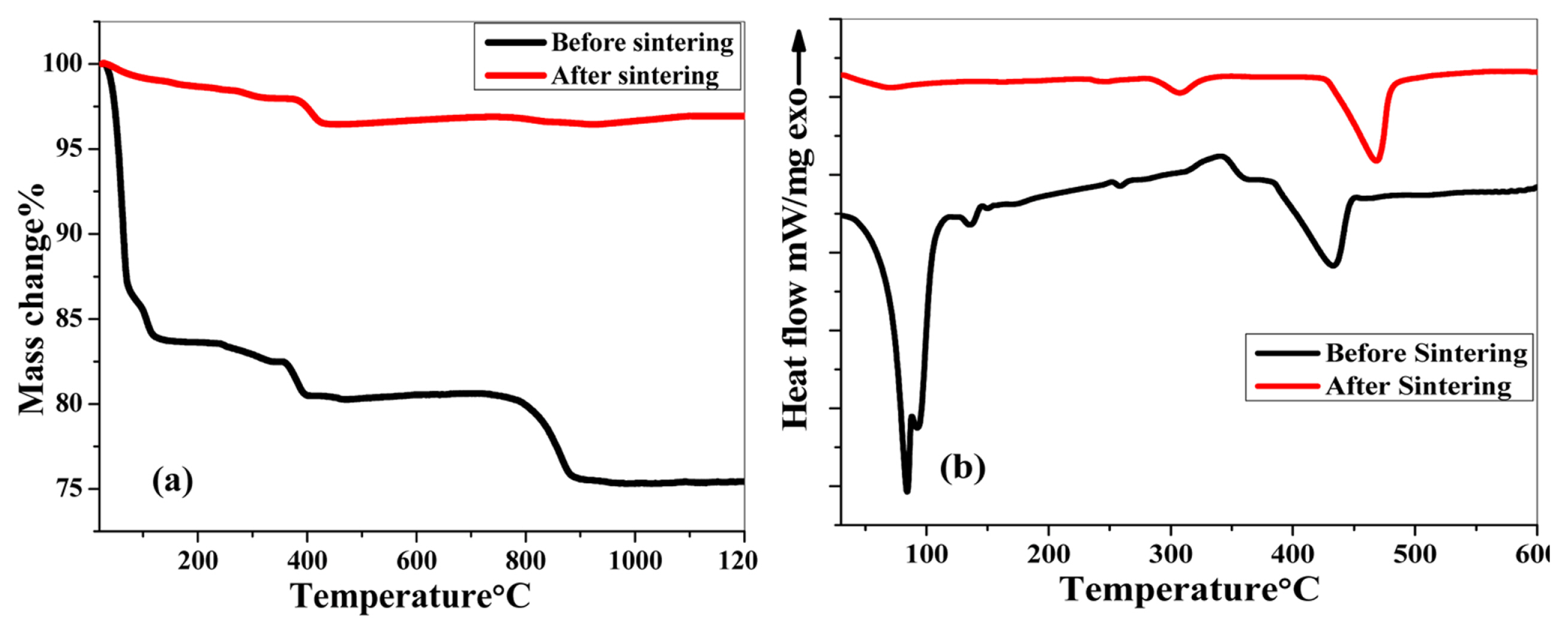
The thermal behavior of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx before and after sintering explained by (a) TGA and (b) DSC.
After sintering the sample at 1150°C followed by air quenching, a total mass loss of only 6% is observed in the region of 382°C to 425°C. The weight loss in this region is attributed to the creation of oxygen vacancies [28]. This negligible weight change suggests that the material possesses high thermal stability and can be easily employed for high-temperature applications.
3.2.2 Differential Scanning Calorimetry
DSC results of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx display two endothermic peaks in the pre-sintered sample as revealed in Fig. 5 (b). The first peak at 83.8°C illustrates the loss of physically adsorbed species on the surface of the oxide as described earlier (Fig. 5 (a)). Before 400°C, most of the residual ions present in the oxide mixture (except carbonates) are either evaporated or volatilized. However, the initiation of the formation of a cubic phase takes place at 433.2°C as depicted by an endothermic peak in this region. While the exothermic peak is associated with the loss of structural water molecules.
From the XRD results of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx in Fig. 3 (b), it became clear that a well-developed spinel structure is formed at 1150°C. The DSC analysis of the HEOx after sintering explains the effect of thermal treatment on the structure of the oxide. Fig. 5 (b) reveals that after sintering two endothermic peaks are observed at a) 307°C and b) 470°C. The first endothermic peak at 307°C is associated with some sort of phase transition within the spinel structure because no weight loss is associated with the TGA curve at this temperature (Fig. 5 (a)). In spinel oxides, the distribution of cations is a function of temperature and pressure and cations distribute themselves in the octahedral or tetrahedral sites by their preference [29]. This redistribution of cations leads to changes in the structure of spinel oxides. However, this endothermic peak is quite small and hence illustrates the stability of the spinel structure after sintering. The second endothermic peak at 470°C arises due to the formation of oxygen vacancies [28]. The creation of oxygen vacancies is quite useful for making the material a better catalyst for various applications. Hence, from the thermal properties of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx, we expect its potential activity in many catalytic processes [30–32].
4. Electrochemical Studies
Electrochemical studies were carried out to evaluate the electrocatalytic properties of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx through cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). CV is the most fundamental technique in electrochemistry which provides rapid assessment about the redox processes, location of their potentials, and the effect of the electrolyte on the processes under study. Diffusion coefficient (Do), heterogeneous rate constant (ko), and different thermodynamic parameters can be evaluated by using this technique. EIS provides the kinetic statistics about the electrode/electrolyte interface with the simultaneous understanding of mass transfer and charge transfer processes (Rct) [33]. Mostly EIS data is presented in the form of the Nyquist plot, which consists of an arc/semicircle if only one process is occurring at the interface. The presence of more than one semicircles in the spectrum is an indication of multiple processes, pointing towards a complex mechanism. This technique is unique in itself because it throws light on the bulk and interfacial properties of the system. Multiple parameters like solution resistance (Rs), Warburg impedance, electron transfer resistance (Rct), and double-layer capacitance can be estimated from a single experiment [34]. From the Nyquist plot, Rs is measured from the intercept of the high-frequency region on the real axis. While the low frequency region is related to the resistances of the charge transfer processes. Double-layer capacitance and Rct explain interfacial kinetics of the electrode. In the real system like us, double layer capacitance is replaced with constant phase element (CPE) because of the irregularities and roughness arising from the nonhomogeneous electrode surface [20]. CPE consists of two sub-parameters (a) capacitance and (b) surface roughness. The value of the surface roughness ranges between zero (when capacitance is equivalent to resistance) to one (capacitance corresponds to an ideal capacitor with a smooth surface).
4.1 Electrochemical response of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx on bare glassy GC electrode
The electrochemical response of bare GC electrode and (Be,Mg,Ca,Sr,Zn,Ni)3O4/GC [later on, in the rest of the text it will be used as (Be,Mg,Ca,Sr,Zn,Ni)3O4] HEOx showed in Fig. 6 (a) is recorded in a potential window of −1.5 V to 1.5 V at a scan rate of 5 mV s−1 in 1 M solution of KOH. The CV results reveal that in the given potential window, HEOx remains completely inert except a small hump is seen in the positive potential window due to the water oxidation reaction. As can be seen from Fig. 6 (a), HEOx shows no peak both in the forward and reverse scan in the potential window that was chosen for the electrooxidation of hydrazine hydrate oxidation i.e. −1.0 V to 0.8 V. Linear sweep voltammetry (LSV) results of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx in Fig. 6 (b) both in the forward and reverse scans complement the results obtained from CV.

(a) Comparison of the electrochemical response of bare GC electrode and (Be,Mg,Ca,Sr,Zn,Ni)3O4/GC HEOx in 1M KOH (b) Linear sweep voltammetry of (Be,Mg,Ca,Sr,Zn,Ni)3O4/GC HEOx in 1 M KOH.
4.2 Hydrazine hydrate oxidation
Cyclic voltammograms in Fig. 7 (a) describe the electrocatalytic behavior of the sample when exposed to different concentrations of the hydrazine hydrate. The concentration effect is investigated with 10 μM to 0.3 mM hydrazine hydrate concentrations in a potential window ranging from −1.0 V to 0.8 V. The onset potential for the oxidation of hydrazine hydrate was observed to be −183.9 mV. Initially, at a small concentration of hydrazine hydrate, the increase in the anodic peak current is very small. While at higher concentrations, not only the anodic peak current is increased but there is a shift towards more positive potential. This is an indication of the irreversible mode of electron transfer process [35]. In aqueous solutions, hydrazine hydrate oxidation proceeds through the formation of several unstable intermediates like N2H4+ [36], N2H5+, and N2H62+ [37]. The whole scheme involves the transfer of four electrons along with the evolution of nitrogen as the end product [36].
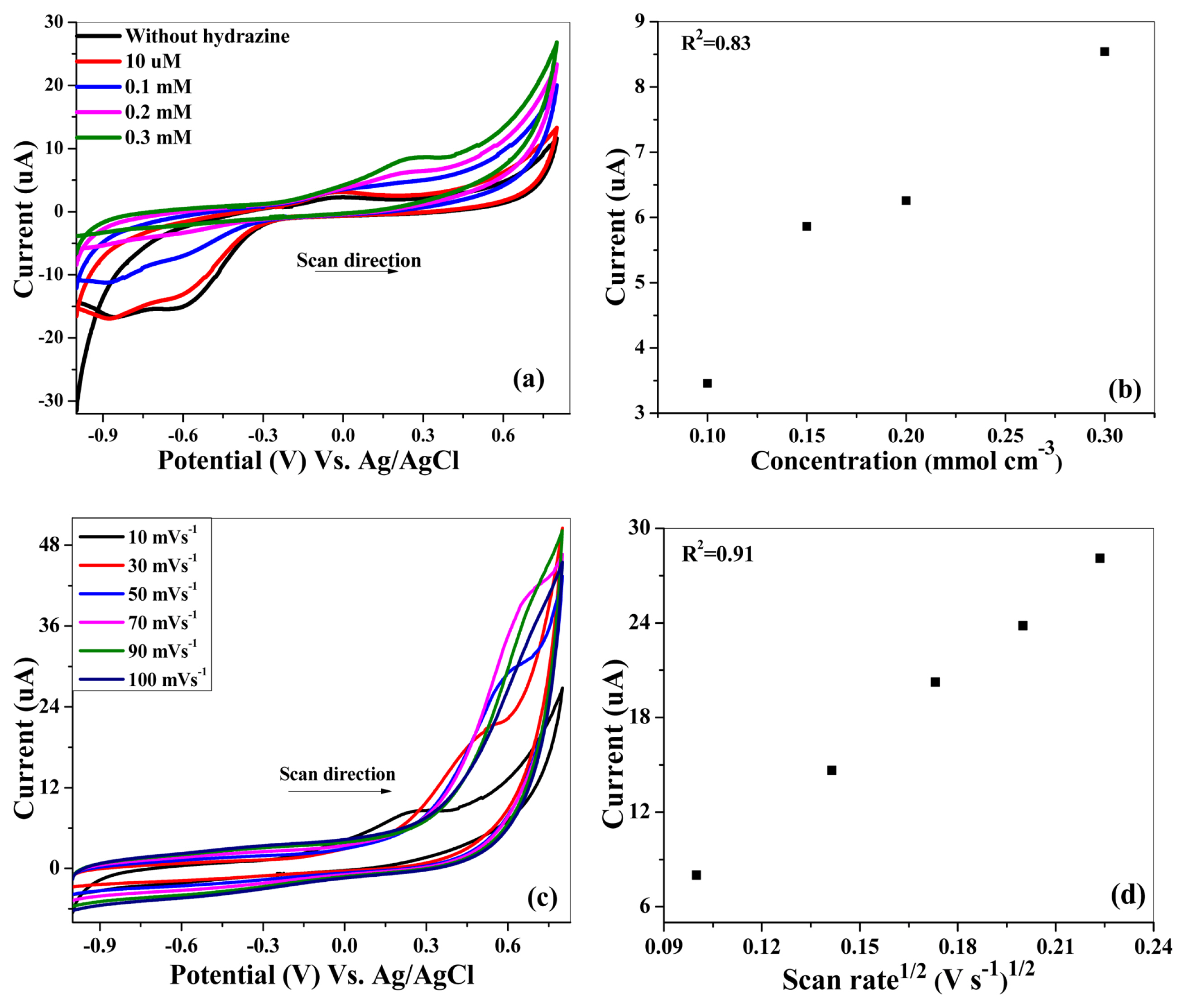
(a) Electrocatalytic oxidation of NH2NH2.H2O on (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx in 0.1 M KOH (b) Straight-line plot of anodic peak current Vs. different concentrations of NH2NH2.H2O in 0.1 M KOH (c) Scan rate effect of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx in 0.1M KOH containing 0.3 mM NH2NH2.H2O (d) Straight-line plot of anodic peak current versus square root of scan rate.
Moreover, the oxygen reduction peak that was appeared in the absence of hydrazine hydrate is disappeared gradually at higher concentrations. It happens as a result of the reaction between hydrazine and the dissolved oxygen (DO), present in the solution. Hydrazine hydrate reacts with the oxygen, lowers the level of DO, and forms nitrogen. However, the rate of this reaction is very low at room temperature because it greatly depends on the temperature and pH of the electrolyte [38].
Fig. 7 (b) illustrates a linear relationship between anodic peak current and concentration of hydrazine hydrate showing the sensitivity of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx towards hydrazine hydrate concentration.
The scan rate effect was studied to understand whether the process of electro-oxidation of hydrazine hydrate is diffusion-controlled or adsorption controlled. Literature studies describe that there are two following ways of this assessment: (a) plotting peak current (Ip) Vs. under root of scan rate (v1/2) i.e., Ip = f (v1/2) or (b) ln Ip = f (ln v). A system (reversible or irreversible) is said to be diffusion controlled when Ip varies linearly with v1/2 [39]. Fig. 7 (c) shows the electrochemical behavior of hydrazine hydrate oxidation by varying the scan rate from 10 to 100 mV s−1 in a solution of 0.3 mM NH2NH2.H2O + 0.1 M KOH. As the scan rate increases, an increase in the anodic peak current takes place accompanied by a shift towards more positive potential. From this, it can be inferred that the understudied process is not only irreversible but also diffusion-controlled as Ipa vs v1/2 varies linearly (Fig. 7 (d)) [35,40]. It points out that the electrooxidation of hydrazine hydrate is directed by the diffusion of hydrazine molecules to the active sites of the catalyst.
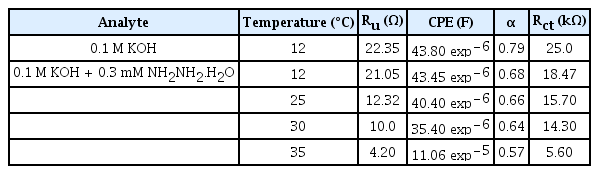
EIS is carried out to get further insight into the kinetics of hydrazine hydrate oxidation both at room and higher temperatures up to 35°C. The catalytic response of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx in 0.1 M KOH and 0.1 M KOH + 0.3 mM NH2NH2.H2O solutions at room temperature (12°C) is shown in Fig. 8 (a). Nyquist plots in Fig. 8 (a) and Table 2 shows that in the presence of hydrazine hydrate, the value of Rct (18.47 kΩ) decreases in comparison to that of its value (25.0 kΩ) in the 0.1 M KOH solution. This fact shows that the (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx has fast electron transfer kinetics for hydrazine hydrate oxidation [41]. While the values of capacitance and Rs are not very much affected by the presence of hydrazine hydrate in the solution at room temperature. Fig. 8 (b) shows the corresponding Bode plots of Fig. 8 (a). It is evident in Fig. 8 (b) that the behavior of catalyst is resistive at higher frequencies as the phase angle is close to zero while the intermediate frequency region shows the presence of reactance in the system [42]. The bode plot in Fig. 8 (b) consists of a onetime constant that indicates the occurrence of the charge transfer process at the electrode/electrolyte interface. When hydrazine hydrate is added to a 0.1 M solution of KOH, a decrease in the phase angle from −66° to −56° is observed along with a shift towards a higher frequency region. These results support the fact that the hydrazine hydrate addition leads to a decrease in charge transfer resistance and fast reaction kinetics [43].
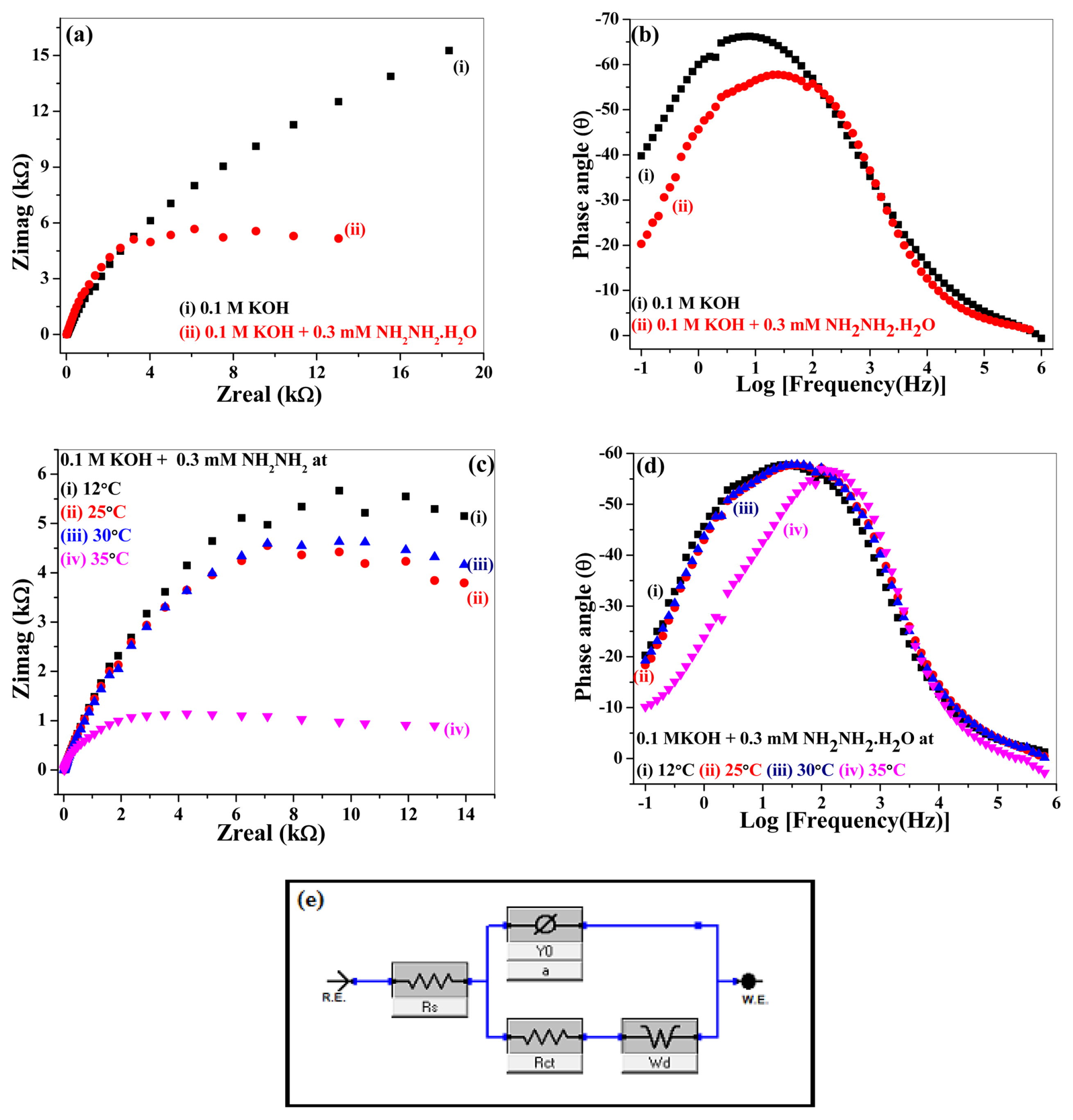
(a) Nyquist plots and (b) Bode plots of (Be,Mg,Ca,Sr,Ni,Zn)3O4 HEOx took in 0.1 M KOH and 0.1 M KOH + 0.3 mM NH2NH2.H2O solutions at room temperature (12°C) (c) Nyquist and (d) Bode plots in 0.1M KOH + 0.3 mM NH2NH2.H2O at (i) 12°C (ii) 25°C (iii) 30°C (iv) 35°C (e) Impedance circuit.
Impedance studies of hydrazine hydrate oxidation catalyzed by (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx in 0.1 M KOH solution at different temperatures.
The effect of temperature on the impedance response and hence kinetics of the hydrazine hydrate oxidation reaction was investigated by recording Nyquist (Fig. 8 (c)) and Bode plots (Fig. 8 (d)). The EIS profiles were recorded in 0.1 M KOH + 0.3 mM NH2NH2.H2O solution at different temperatures i.e., (i) 12°C (ii) 25°C (iii) 30°C and (iv) 35°C. (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx shows analogous catalytic behavior except at 35°C whereby a tremendous increase in the value of CPE is observed. During the electrochemical process, the elevated values of capacitance or CPE are indicative of the higher concentration of charged species present on the electrode surface [44]. As a result of this, at 35°C, a shift towards higher frequency is observed as explicit by Bode plot in Fig. 8(d) indicating a decrease in the resistance complemented by a small semicircle shown by the Nyquist plot in Fig. 8(c).
As can be seen from Table 2, the values of Rs (4.20 Ω) and Rct (5.60 kΩ) at 35°C are exceptionally very small as compared to that of their values at low temperatures. These results show that the potential of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx catalyst to catalyze the oxidation of hydrazine hydrate at higher temperatures is significantly improved due to fast reaction kinetics of the system. As the temperature goes on, the value of α decreases which corresponds to the rough surface of the electrode. Smaller the magnitude of α, more rough is the surface of the electrode [44].
4.3 Water Oxidation
The electrocatalytic potential of (Be,Mg,Ca, Sr,Zn,Ni)3O4 HEOx towards water oxidation is evaluated by varying the scan rate from 20 mV s−1 to 100 mV s−1 in a potential window ranging from 0 V to 1.7 V in 0.1 M KOH solution as shown in Fig. 9 (a). The onset potential for water oxidation reaction is observed to be 0.218 V. The linear relationship between Ip and v1/2 in Fig. 9 (b) shows that the water oxidation on the (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx catalyst surface is diffusion controlled.

(a) Cyclic voltammetric response of (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx for water oxidation at different scan rates (b) Straight-line plot of anodic peak current and the square root of scan rate (c) Nyquist and (d) Bode plots explaining electrode/electrolyte interface processes (e) Impedance circuit.
EIS is executed to probe the detailed kinetics of the electrode/electrolyte interface during water oxidation. The circuit diagram for the water oxidation process in Fig. 9 (e) contains a solution resistance (Rsoln) and a double layer capacitance of the electrode (Cdl) [45]. While Rp and Rs are the resistances associated with the interfacial charge transfer reaction and C is the capacitance in parallel with Rs represents the parameters related to the relaxation of the adsorbed intermediates during the process of water oxidation [44]. The Nyquist plot in Fig. 9 (c) shows two depressed semicircles corresponding to two-time constants observed in the Bode plot as shown in Fig. 9 (d). While looking at Fig. 9 (d), it becomes clear that at high and low frequencies, the electrochemical system faces maximum resistance as the phase angle is close to zero. In the intermediate region of the frequency, the first hump that appears around 103 Hz is related to the processes occurring at the electrode/ electrolyte interface. This is associated with the double-layer capacitance and charge transfer of the intermediate products formed during the process (interfacial charge transfer reaction) [46,47]. The physical meaning of double-layer capacitance is that it shows the extent to which the electrode area is in contact with the electrolyte [48]. The impedance profile of HEOx at a low frequency regime (near 101 Hz) in Fig. 9 (d) is associated with the evolution reaction of oxygen upon the surface of metal oxides from the reaction centers involved in water oxidation process [47].
4.4. Comparison of Catalytic Potential in Water Oxidation and Hydrazine Hydrate Oxidation
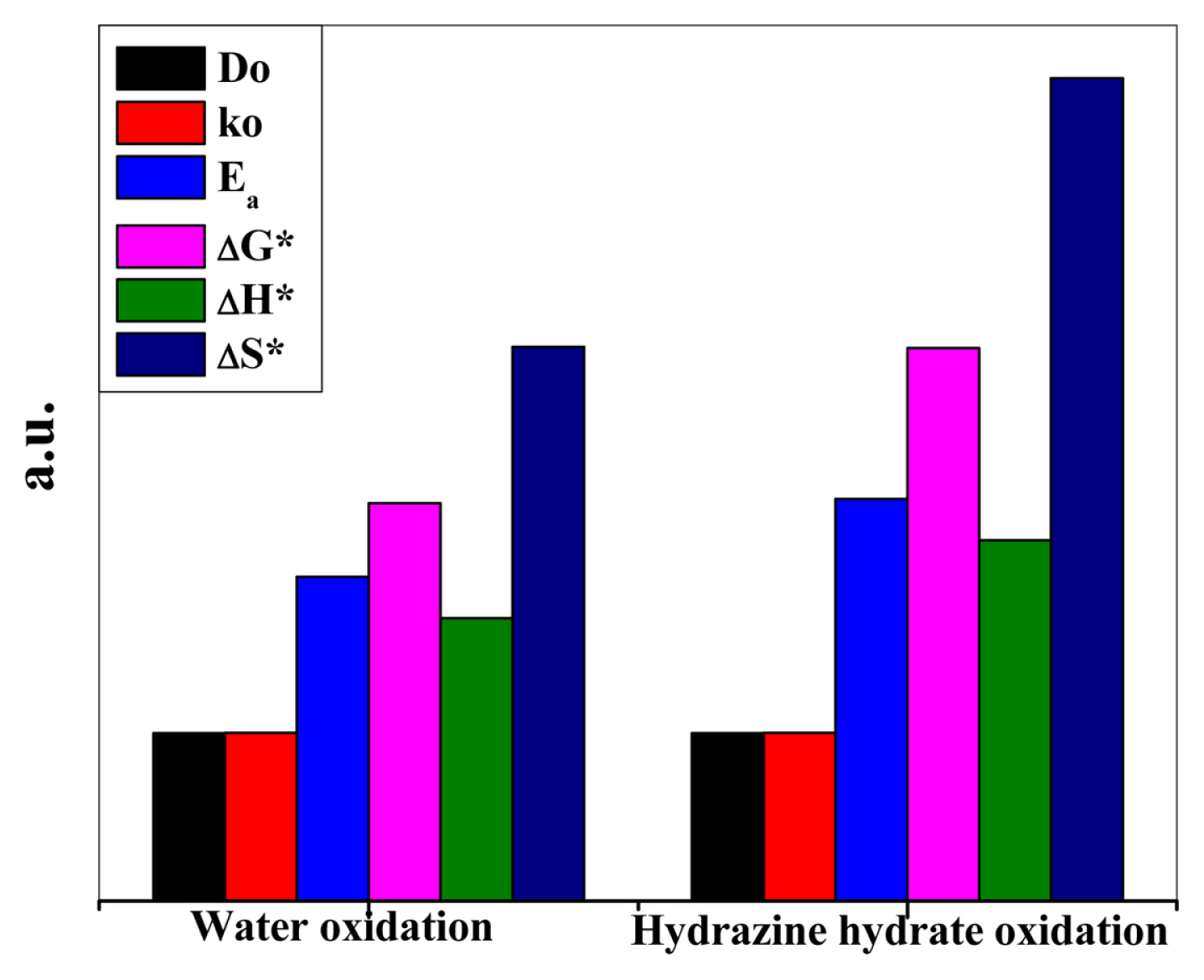
The electrochemical results shown in Table 3 reveal that (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx has the potential to catalyze the oxidation of both water and hydrazine hydrate. Both of these reactions are very important regarding their application in the fuel cells. Bar graph in Fig. 10 shows a comparison of the kinetic and thermodynamic parameters of water and hydrazine hydrate oxidation at room temperature at a scan rate of 20 mV s−1 (Yaxis is labelled as a.u. because it represents a comparison of the catalytic activity of the (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx towards water and hydrazine hydrate oxidation based on different kinetic and thermodynamic parameters). For irreversible systems, kinetic parameters Do and ko were calculated from the following equations
Kinetic and thermodynamic parameters for hydrazine hydrate and water oxidation.
Comparison of thermodynamic and kinetic parameters of the water and hydrazine hydrate oxidation.
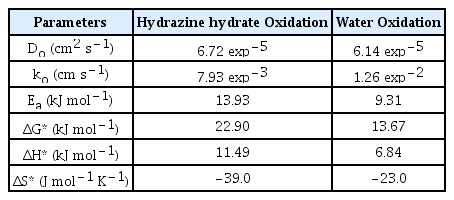
Where Ip is the anodic current, F, A, and C are the Faraday constant, geometric area of the electrode, and concentration of the reacting species (hydrazine hydrate and oxygen) respectively. While α is the transfer coefficient, n is the total number of electrons, and nα is the number of electrons involved in the rate-determining step. Table 3 shows that the Do (6.14 exp−5 cm2 s−1) and ko (1.26 exp−2 cm s−1) values for water oxidation are higher as compared to that of hydrazine hydrate oxidation. Hence, (Be,Mg,Ca, Sr,Zn,Ni)3O4 HEOx catalyzes the oxidation of water more efficiently as compared to that of hydrazine hydrate oxidation.
To evaluate the thermodynamic parameters, following mathematical relationship based on Marcus theory is used [35].
Zhet is the collision number for any heterogenous electron transfer reaction and can be calculated from the following equation.
Here, M is described as the molecular mass of the hydrazine hydrate in hydrazine oxidation and oxygen for water oxidation. The value of Gibbs free energy (ΔG*) determines the feasibility of the chemical reaction from the following relation between enthalpy (ΔH*) and entropy (ΔS*) of the reaction.
From ΔH*, activation energy (Ea) of the reaction can be estimated by using the following relation [49].
From a closer look at the thermodynamic parameters (Table 3), it can be inferred that the catalyst is more sensitive towards water oxidation. It is because the Ea required to accomplish the hydrazine hydrate oxidation is 13.93 kJ mol−1 while its value comes out to be 9.31 kJ mol−1 for water oxidation. Moreover, the smaller values of ΔG* (13.67 kJ mol−1) and ΔH* (6.84 kJ mol−1) makes the HEOx a more suitable catalyst for water oxidation as compared to hydrazine hydrate oxidation. Positive values of ΔG* and ΔH* indicate that the electrooxidation of both the processes is non-spontaneous and activation controlled. While the negative values of ΔS* illustrate the adsorptive nature of the products formed during the reaction.
The catalytic potential of the equiatomic nanocrystalline (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx towards the electrooxidation of water and hydrazine hydrate was explored through CV and EIS. These electrochemical studies clearly showed that the catalyst has the potential to oxidize both water and hydrazine hydrate. The electrochemical results in Figs. 7 (c) and 9 (a) illustrated that the process of hydrazine hydrate and water oxidation (respectively) is irreversible and diffusion controlled. However, the Nyquist plot in Fig. 9 (c) showed that at the electrode/electrolyte interface the charge transfer resistance for water oxidation is lower as compared to that for hydrazine hydrate oxidation, as depicted in Fig. 8 (a). This aspect revealed that the process of water oxidation has fast kinetics than that of the hydrazine hydrate oxidation on the surface of the catalyst. Moreover, a comparative study of the thermodynamic and kinetic parameters in table 3 also illustrated that (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx has better catalytic response towards water oxidation than hydrazine hydrate oxidation. Because the former has a lower activation energy (Ea) and higher rate constant (ko).
5. Conclusions
A novel equiatomic hexanery (Be,Mg,Ca,Sr, Zn,Ni)3O4 HEOx was successfully synthesized by a beautiful blend of alkaline earth and 3d transition metal oxides. In its synthesis, the solid-state reaction route was adopted followed by sintering and air quenching to room temperature. Following are the findings:
Structural properties reconnoitered by XRD and SEM analyses gave insight into the crystal structure and microstructure of HEOx understudy respectively. XRD confirmed that the (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx possesses a single-phase with Fd-3m spinel structure and the crystallite size in the nanometer regime. SEM results show that the HEOx exhibits irregular morphology and the agglomerations of nanoparticles. It is attributed to the fact that the air quenching locks the crystal structure and particle size of compounds at high temperatures. Hence, (Be,Mg, Ca,Sr,Zn,Ni)3O4 HEOx is, in fact, nanocrystalline.
Thermal properties of sintered and air quenched (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx explored by TGA and DSC revealed that it possesses excellent thermal stability showing no major weight loss or weight gain in TGA curve up to the studied temperature of 1200°C. Moreover, the DSC curve shows the creation of oxygen vacancies around 470°C making it a potential candidate to be used as a catalyst especially in water oxidation, and hydrazine hydrate oxidation, etc.
Electrochemical applications of (Be,Mg,Ca, Sr,Zn,Ni)3O4 HEOx studied through CV gave an insight into its potential to catalyze both water and hydrazine hydrate oxidation. Among kinetic parameters, the values of Do (6.14 exp−5 cm2 s−1) and ko (1.26 exp−2 cm s−1) are higher for water oxidation as compared to hydrazine hydrate oxidation (Do = 6.72 exp−5 cm2 s−1, ko = 7.93 exp−3 cm s−1). Moreover, the comparative thermodynamic values for water oxidation (ΔG* = 13.67 kJ mol−1, ΔH* = 6.84 kJ mol−1) and hydrazine hydrate oxidation (ΔG* = 22.90 kJ mol−1, ΔH* = 11.49 kJ mol−1) also reflect that electrocatalysis of water oxidation is more facile at room temperature. Hence, both kinetic and thermodynamic values depict that the water oxidation is more facile on (Be,Mg,Ca,Sr,Zn,Ni)3O4 HEOx surface as compared to hydrazine hydrate oxidation.
Acknowledgments
Authors gratefully acknowledge the financial support provided by the Higher Education Commission (HEC) of Pakistan under the “Access to Scientific Instrument Program (ASIP)” scholarship through the award letter number: 20-2(10)/ASIP/R&D/HEC/17/ 000619(IST, Ibd)/26. Authors would like to thank Dr. Mujtaba Shah and Dr. Amin Badshah for providing lab facilities.
Notes
Conflicts of interest
The authors declare no conflicts of interest.