Challenges and Design Strategies for Conversion-Based Anode Materials for Lithium- and Sodium-Ion Batteries
Article information
Abstract
Although lithium-ion batteries are currently the most reliable power supply system for various mobile applications, further improvement in energy density is still required as the need for batteries in large energy-consuming devices is rapidly growing. However, in the anode, the most widely commercialized graphite-based anode materials almost face theoretical limitations. In addition, sodium-ion batteries have been actively studied to replace expensive charge carriers with cheaper ones. Accordingly, conversion-based materials have been extensively studied as high-capacity anode materials in both lithium-ion batteries and sodium-ion batteries because their theoretical capacity is twice or thrice higher than that of insertion-based materials. This review will provide a comprehensive understanding of conversion-based materials, including basic charge storage behaviors, critical drawbacks that should be overcome, and practical material design for high-performance.
1. Introduction
Globally, tremendous efforts have been devoted to changing the power source from fossil fuels to renewable energy due to environmental issues. Thus, increasing the energy density of batteries is becoming increasingly significant for stably operating smart grids and manufacturing long-distance electric vehicles. Currently, lithium-ion batteries (LIBs) exhibit the most reliable and superior performance among various batteries considering energy and power density, life span, and reduced self-discharging [1–3], resulting in an enormous success of LIBs dominating the market share. However, the current performance of LIB technology remains insufficient as power sources for energy grids and electric vehicles, owing to the limited capacity of commercial electrodes. Therefore, researchers are attempting to develop high-capacity electrode materials for improving the energy density of LIBs. In addition, concerns regarding the availability of lithium sources have led to the research interest in developing new energy storage techniques using earth-abundant elements. Sodium-ion batteries (SIBs) are one of the promising energy storage systems for large-scale stationary energy grids that can reduce the burden of LIBs due to sufficient sodium sources [4,5]. Although various materials have been applied as electrode materials for SIBs so far, their performance is not yet appropriate for commercial use.
To date, insertion-based materials such as layered transition metal oxides and graphite have been most widely used in commercial LIBs. Generally, approximately 1 mol of lithium-ion can be stored per formula unit based on the insertion reaction mechanism, as insertion-based materials accommodate lithium-ions only at the specific insertion site [6–9]. Thus, only a limited capacity can be achieved using insertion-based materials. Particularly for the anode, conventional graphite materials almost meet the theoretical limits [10]. Accordingly, conversion-based materials have been extensively researched as potential candidates for replacing commercial graphite anode materials to surpass the limitations of current LIBs [11,12], since the first report in the early 2000s [13]. In the first report, P. Poizot et al. observed that the nanosized transition metal can store plenty of lithium-ions by the following conversion reaction mechanism; MxOy + 2yLi+ + 2ye− ↔ xM0 + yLi2O. During discharging, the transition metal is completely reduced to the metallic phase with the formation of Li2O. At subsequent charging, the Li2O phase is reversibly decomposed, and M-O bond is formed. The theoretical capacity of conversion-based metal oxides is much larger, exceeding 500–1000 mAh g−1. In addition to oxides, various metal compounds undergo conversion reactions exhibiting large specific capacity [14,15]. As sodium exhibits similar chemical properties to lithium in various aspects, numerous conversion-based materials have also been applied as anode materials for SIBs [16–18]. Identical to LIBs, conversion reaction mechanisms can be observed in various metal compounds, delivering higher reversible capacity than insertion-based materials.
Despite large theoretical capacity, conversion-based anode materials remain far from commercialization. Owing to the severe volume changes during electrochemical cycling and the sluggish reaction kinetics, the cycle and rate capability are relatively poor compared to those of insertion-based materials. Low energy efficiency is another severe drawback of conversion-based materials. For the realization of commercial conversion-based anode materials with high energy density, innovative advances should be established for overcoming the obstacles.
This review will first address basic charge storage behaviors of conversion-based materials, including detailed ion storage mechanisms and microstructural changes. The recent developments in various types of conversion-based materials are then summarized. Finally, critical issues that hinder commercialization and some effective strategies for enhancing the electrochemical performance will be provided.
2. Charge storage behaviors of conversion-based anode materials
2.1. Ion storage reaction mechanism
For detailed identification of the ion storage reaction mechanism, various analytical techniques were utilized to observe changes in the crystal structures, electronic states, and bonding characteristics during electrochemical cycling [19–21]. Among them, X-ray diffraction (XRD) is an efficient technique for investigating the crystallographic structure with long-range order. X. Ou et al. inspected the structural changes of NiSe2 anode materials for SIBs using in situ XRD experiment. During discharge to ~1.1 V, the peaks from NiSe2 shift toward a lower angle due to lattice expansion from sodium-ion insertion to form NaxNiSe2 phase. At lower potential, the Nax− NiSe2 peaks decreased, and new peaks from Na2Se phase grew, demonstrating the occurrence of the conversion reaction. During subsequent charging, the Na2Se peaks were gradually reduced, and NiSe2 peaks were observed again after complete charging, indicating the reversibility of the conversion reaction [22]. Similarly, other researchers have proven the conversion reaction mechanism using XRD techniques by observing the metallic phase and LiX/NaX phases after full discharge [23–28]. However, although XRD is a powerful method for obtaining phase-sensitive information, it is not always the best choice for identifying conversion reactions because most conversion-based materials significantly lose their crystallinity during lithiation.
Unlike XRD, X-ray absorption spectroscopy (XAS) probes the chemical state and local structure of target elements in short-range order. Thus, it is more suitable for observing changes occurring at the nanoscale to explore structure changes during conversion reaction [29–33]. J. Yoon et al. investigated the charge storage behavior of mesoporous MnO2 using Mn K-edge and L-edge XAS [34]. As shown in Mn K-edge Fourier-transformed extended X-ray absorption fine structure (EXAFS) in Fig. 1(a)–(e), the initial MnO2 peaks gradually decrease, and new peaks appear at approximately 1.6 and 2.6 A, indicating the phase transition of MnO2 to MnO phase. Further lithiation leads to the formation of metallic Mn, as indicated by the peak at ~2.2 A that attributed to Mn-Mn interaction in Mn metal. At the last stage of discharge, the Mn metal peak only slightly increases, although a large capacity of ~600 mAh g−1 was exhibited. The authors insist that this capacity originates from the formation of the electrolyte-derived surface layer. Fig. 1(f) shows that the peaks at ~642 and 652 eV Mn L-edge XANES disappear after discharge and appear again during charge. As the total electron yield mode is a surface-sensitive experiment, the Mn L-edge XANES result demonstrates that the electrolyte-derived surface layer is formed during discharge reversibly decomposed during charge. During charging, the Mn-Mn interaction from Mn metal decreases, and Mn-O bonding at ~1.6 A from the MnO phase appears again without long-range ordering. Then, the Mn-O interaction peak at ~1.6 A decreases gradually and increases again at the slightly lower position. Simultaneously, the Mn-Mn bond peak at ~2.3 A grows. The EXAFS spectra during charge indicate that the Mn metal formed by conversion reaction initially converted to amorphous MnO and further oxidized to Mn3O4.

Additionally, transmission electron microscopy (TEM) [35–38], X-ray photoelectron spectroscopy (XPS) [39–42], pair distribution function (PDF) [43–45], and other tools have also been used to scrutinize precise charge storage mechanisms. As a result of extensive research, the typical ion storage behaviors of conversion-based anode materials have been reported as follows. First, the insertion reaction occurs if there are available sites for cation accommodation inside the crystal. Then, conversion reaction occurs to form extremely small-sized metal and LiX/NaX phases. Finally, the capacity is delivered without changing the electronic and local structures of active particles in the low potential region, which is generally called extra capacity. This results in a high capacity over the theoretical value, as in the mesoporous MnO2 [34]. This extra capacity is attributed to abnormal charge storage reactions [11,46,47] such as electrolyte-derived surface layer [48], interfacial charge storage [49,50], reaction of lithium-containing species [43,51], and/or ion storage in defects/metallic lithium storage [52,53]. During subsequent charging, changes occur in the opposite manner. The reversible abnormal charge storage reaction occurs first, and the metallic phase is then oxidized at a relatively high potential.
2.2. Microstructural changes during conversion reaction
Unlike insertion-based materials where the initial frameworks are relatively well maintained during electrochemical cycling, morphology of conversion-based materials changes significantly. Such microstructural variations in conversion-based anode materials can be monitored in real-time with atomic resolution thanks to the significant advances in TEM techniques, enabling in situ experiments [35,36,54,55].
K. E. Gregorczyk et al. investigated the microstructural changes of RuO2 nanowires using in situ TEM [56]. The TEM images in Fig. 2(a)–(d) show that the nanowire diameter expands sequentially, starting from the region near the lithium sources. The diameter notably increases by ~41% after lithiation, while the length increases slightly. Totally, lithiation results in a large volume expansion of ~95%. In addition, the initial single-crystalline rutile RuO2 crystal as shown in Fig. 2(e) transformed into polycrystalline mixture of metallic Ru and Li2O (Fig. 2(f)). The magnified view near the reaction front in Fig. 2(g) clearly shows the contrasting nature of the crystalline initial RuO2 crystal and the reacted side. On the reacted side, extremely small metallic Ru nanoparticles are visible in the Li2O matrix, as shown in Fig. 2(h).
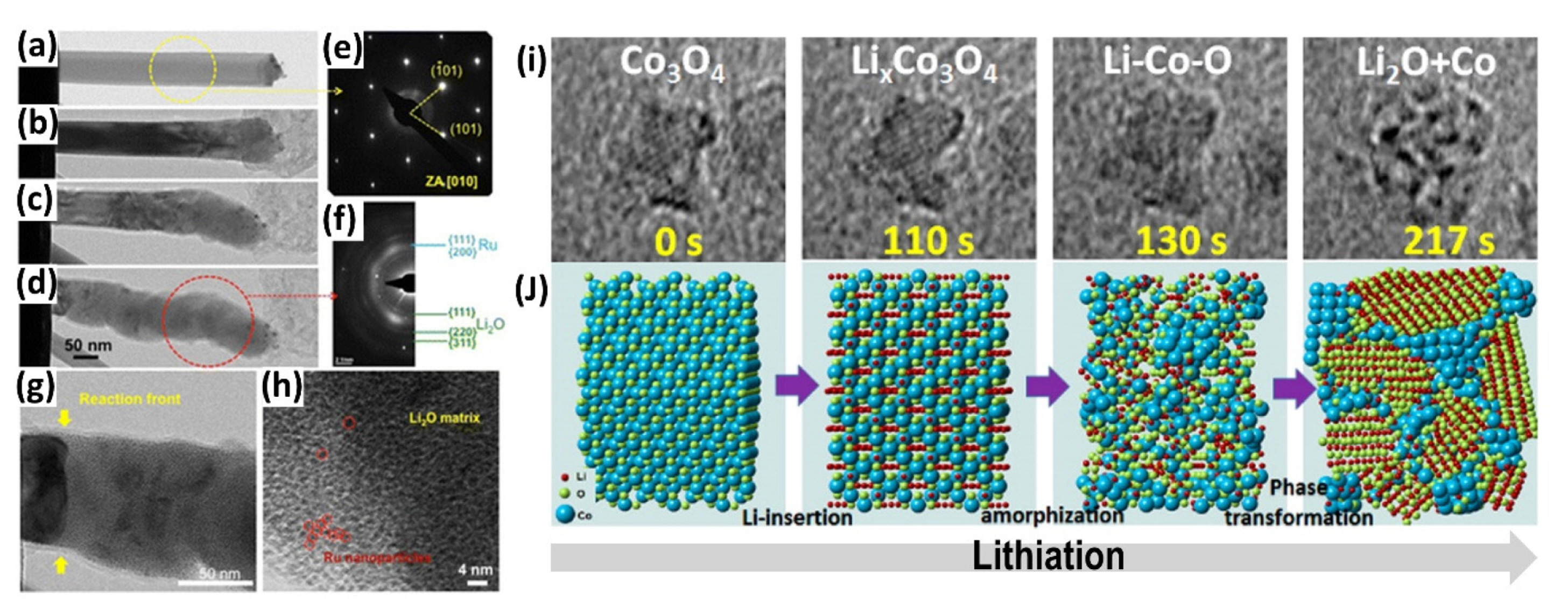
(a)–(d) in situ TEM images during the first lithiation step of RuO2 nanowire according to the reaction time. Electron diffraction patterns of (e) pristine and (f) lithiated RuO2. Magnified TEM images (g) near the reaction front and (h) of the lithiated region. (a)–(h) Reproduced with permission from Ref. [56] Copyright © 2013 American Chemical Society. (i) in situ TEM images recorded during lithiation of Co3O4 nanocubes on few-layer graphite with (g) corresponding schematic atomistic models. (i),(g) Reproduced with permission from Ref. [57] Copyright © 2014 American Chemical Society.
L. Luo et al. also conducted in situ high-resolution TEM experiment with Co3O4 nanocubes grown on few-layer graphene [57]. As shown in Fig. 2(i), the Co3O4 nanocubes slightly expanded by 14 % in the 2D area after 110 s, with the original crystal framework remaining unbroken. In this stage, the lithium-ion is inserted into the crystal. During further lithiation, the crystalline nature collapses into a nearly amorphous state, and the particle expands by ~25%. At the last stage of lithiation, the volume significantly expanded to ~55% in the 2D area. In addition, the Co metal phases are formed at 3–5 nm, which appear as dark clusters in the TEM images, embedded in amorphous Li2O nanocrystals. The lithiation behavior of Co3O4 investigated by in situ TEM is summarized in Fig. 2(j). Initially, intermediate LixCo3O4 and nano-sized Co-Li-O phases are formed before the conversion reaction. At full discharge, Co metal clusters are formed and surrounded by the Li2O phase.
Together with these microstructural changes in the active particles, the surface of electrode changes extensively. S. Grugeon et al. discovered a formation of thick amorphous layer after discharging Cu2O electrode to 0.02 V, which then disappeared during subsequent delithiation [58]. S. Laruelle et al. first named this amorphous layer a polymeric/gel-like film (PGF) to differentiate it from the indecomposable solid electrolyte interphase (SEI) layer. They assumed that the formation/decomposition of the PGF accounts for the extra capacity over the theoretical value [48], as mentioned in the above section. However, distinguishing SEI and PGF remains unclear because the components are similar in both cases, and there are controversial arguments about decomposable components [40,41,59,60]. As the components of the surface layer such as lithium/sodium carbonate, fluoride, alkyl carbonate, and hydrocarbons are decomposition products of the electrolyte, the term electrolyte-derived surface layer is used [46,61–63] along with the PGF and SEI.
The ion storage reaction mechanism and morphological changes during electrochemical cycling in conversion-based materials are schematically illustrated in Fig. 3. After discharge, the initial crystal framework collapsed into a nanosized metal embedded in the LiX/NaX phase with a thick amorphous electrolyte-derived surface layer. During charge, deconversion reaction occurs with the decomposition of the surface layer. However, the crystalline nature does not recover to the initial state even after full delithiation.
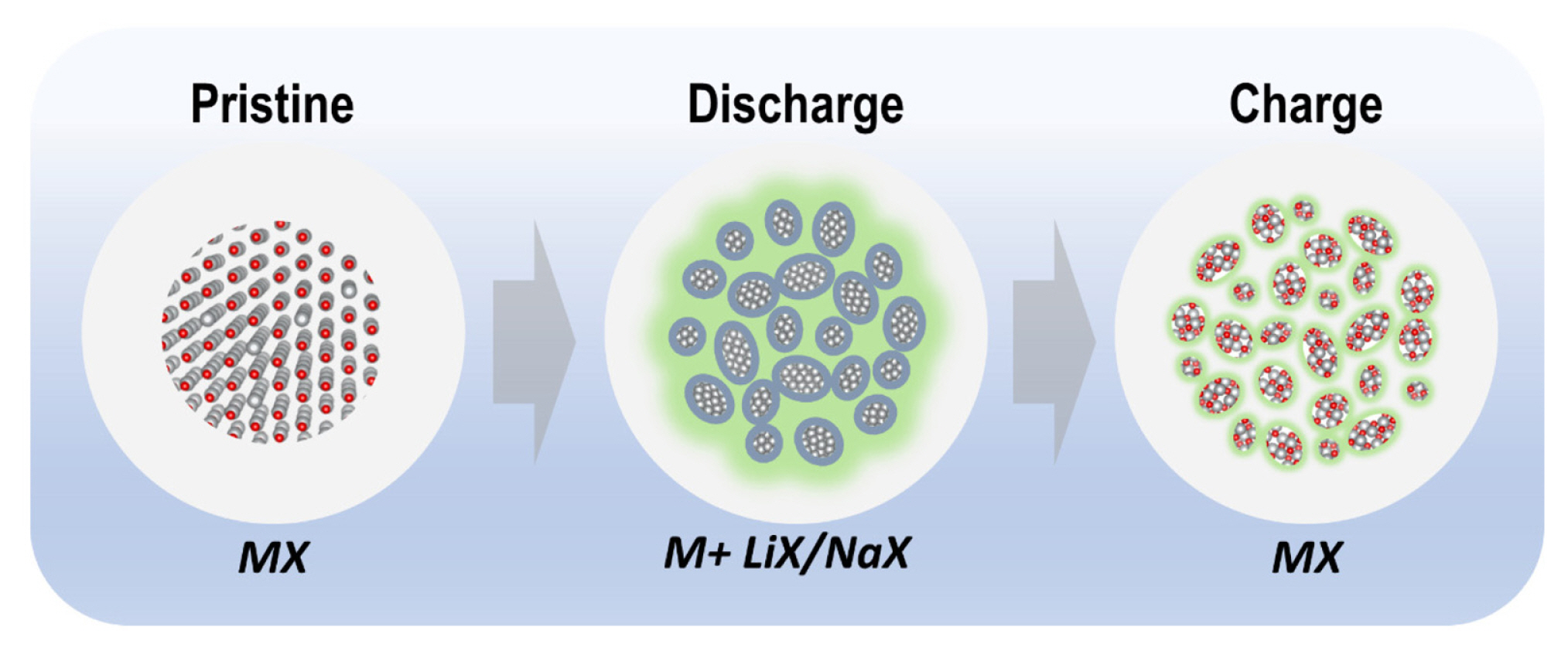
Schematic illustration of crystallographic and microstructural changes of conversion-based materials during electrochemical cycling.
3. Conversion-based metal compounds
A variety of metal compounds store electrochemical energy via conversion reactions. The theoretical and reported practical capacity of some conversion-based anode materials are listed in Table 1. Despite the identical theoretical capacity, the practical capacity of conversion-based materials is generally lower in SIBs than in LIBs due to the slow kinetics of sodium-ions.

Specific capacity of various reported conversion-based anode materials at the first discharge and charge process, and after long-term cycling (unit: mAh g−1).
3.1. Oxides
In 2000, P. Poizot et al. reported the reversible electrochemical reactivity of nanosized transition metal oxides toward lithium-ions [13]. Unlike the conventional view of inactive Li2O, the electrocatalytic properties of metal nanoparticles and nanosizing activate the reversible decomposition of Li2O, enabling reversible conversion reaction.
Since then, various conversion-based metal oxides (metal: Cr, Mn, Fe, Co, Ni, Zn, Sn, Mo, Ru, W, etc.) have been extensively tested as anodes for LIBs, owing to their high theoretical capacity and relatively easy preparation methods. These materials often exhibit considerably high gravimetric and volumetric capacity [14,18,98]. Manganese oxides (MnO, Mn3O4, Mn2O3, MnO2, etc.), for example, have been widely studied as anode materials for LIBs owing to their low cost, environmentally benign nature, and high theoretical capacity [30,34,99]. The theoretical capacity depends on the oxidation state of Mn, but the charge storage reactions are quite similar. During lithiation, insertion reaction occurs first, and then, intermediate MnO phase can be observed. At full discharge, metallic Mn and Li2O phase can be observed, which is typical behaviors of conversion-based anode materials. During charge, metallic Mn is oxidized to Mn2+ at ~1.3 V and further oxidized to a higher oxidation state at over ~2.3 V [30,34,99]. In case of RuO2, lithium is first inserted into rutile structures. Then, conversion reaction occurs at ~0.8 V plateau, forming nanosized Ru metal and Li2O. At low potential slope region, capacity is delivered by abnormal charge storage reaction, and the detailed mechanism is still debated [43,100]. Due to the contribution of the additional abnormal reactions, the reversible capacity exceeds the theoretical value.
The metal oxides are also attractive anode materials for SIBs. Although the reversible capacity is generally much lower in SIBs, it is still much larger than that of insertion-based anode materials. In addition, the lower operating potential of conversion reaction in sodium-based systems is beneficial as anode materials [101,102]. Thus, various oxide materials have been adopted as anode materials, as in the case of LIBs [70,71,103,104]. S. Hariharan et al. compared the electrochemical performance of Fe3O4 anode materials for LIBs and SIBs [103]. In LIBs, high reversible capacity of ~950 mAh g−1 was delivered. However, ~366 mAh g−1 was exhibited because of the sluggish kinetics of sodium-ions, as the ionic radii of Na+ are larger than Li+. M. Xu et al. synthesized hollow spheres of carbon-confined Co3O4, which exhibited high reversible capacity of ~700 mAh g−1 as anode materials for SIBs and retained ~74.5% of capacity after 500 cycles [70].
3.2. Other chalcogenides
Although the theoretical capacity of metal chalcogenides is lower than that of oxides due to their heavier molecular weight, they occasionally surpass the electrochemical performance of metal oxides. Compared to oxides, metal chalcogenides have better intrinsic electrical conductivity and weaker metal-ligand bond strength, leading to faster reaction kinetics and high reversibility [105–107]. Furthermore, the discharge product of sulfides (Li2S/Na2S) is more conductive than oxides (Li2O/Na2O) [108,109]. In addition, the unique properties of tunable stoichiometric and rich redox sites of metal chalcogenides have attracted much interest [107,110]. J. S. Cho et al. show that FeS nanofibers exhibit higher sodium-ion storage capacity of ~456 mAh g−1 while hollow Fe2O3 delivers ~328 mAh g−1 at a constant specific current of 500 mA g−1, although the theoretical capacity of FeS (610 mAh g−1) is much lower than that of Fe3O4 (1007 mAh g−1) [76]. Similarly, G.-L. Xu et al. reported higher sodiation performance of Co3S4 than that of Co3O4 [111]. In LIBs, some metal sulfides also exhibit exceptionally high capacity over ~1000 mAh g−1 [112,113].
Likewise, many metal selenides and tellurides undergo conversion reaction during lithiation and sodiation, forming Li2Se/Na2Se or Li2Te/Na2Te with metallic phases. Some of these materials exhibit stable performance during high-rate cycling despite relatively lower capacity due to heavier elements [82,114–117].
3.3. Nitrides and phosphides
During lithiation of metal nitrides, the metal atom is reduced to metallic phases embedded in the Li3N matrix, which is a lithium superionic conductor. Thus, metal nitrides are attracting much interest as anode materials for LIBs owing to the beneficial properties of conversion reaction products and intrinsically high electronic conductivity [118,119]. G. Jiang et al. observed conversion reaction mechanism forming Co metal and Li3N phase after irreversible insertion reaction in ordered mesoporous CoN anode, delivering high reversible capacity of ~889 and ~616 mAh g−1 at a specific current of 0.2 and 1.0 A g−1 [83]. X. Li et al. confirmed the high and stable reactivity of FeN@C hollow nanocubes as anode materials for both LIBs and SIBs, even at high specific current of 5 A g−1 [120].
High theoretical capacity and earth-abundant phosphorous make metal phosphides also attractive as anode materials. Metal phosphides undergoes both conversion and alloying reaction. Thus, theoretical capacity of some 3d-transition metal phosphides exceeds ~1000 mAh g−1 (MnP4: ~1800 mAh g−1, CoP3, NiP3: ~1590 mAh g−1).
J. Fullenwarth et al. confirmed the reversible conversion reaction of NiP3 in both LIBs and SIBs. The NiP3 exhibits high reversible capacity of ~1441 mAh g−1 as an anode material for LIBs and ~1022 mAh g−1 in SIBs at a C/10 rate [121]. K.-H. Kim et al. reported the superior cation storage performance of MnP4 nanoparticles. Significantly high capacity of ~1600 and ~1028 mAh g−1 was delivered in LIBs and SIBs, respectively, by the conversion and alloying reaction forming metallic Mn and Li3P/Na3P phases [122].
3.4. Polyanionic compounds
In lithium-based system, various types of polyanionic metal compounds such as hydroxides, sulfates, carbonates, and oxalates undergo conversion reaction [51,90,93,123,124]. According to the conversion reaction mechanism, the theoretical capacity of these materials is lower than that of oxides with the same oxidation state of metal atoms because of their heavier molecular weight. However, in many cases, the actual reversible capacity far exceeds the theoretical value, which cannot be explained by the formation of electrolyte-derived surface layer and interfacial charge storage reaction as these reactions generally contribute to only ~200–400 mAh g−1. Advanced analytical techniques demonstrate that the additional capacity originates from the reversible redox reaction of lithium-containing species such as LiOH, Li2CO3, LiSO4, and Li2C2O4, which are the conversion reaction products [46,51,90,93,125,126]. In 2013, L. Su et al. observed the Li2O phase and low-valence C after lithiation of CoCO3, in addition to the conversion reaction products of Co metal and Li2CO3. They suggested that the additional reaction mechanism of reducing C4+ in Li2CO3 into low valence C with the formation of Li2O, exhibiting extra capacity over theoretical expectations [90]. Owing to the additional redox reaction of Li2CO3, some metal carbonates even exhibit higher practical reversible capacity than the oxides that have the same oxidation state, despite the lower theoretical capacity [127]. Additional ion storage mechanism of conversion reaction products was also observed for Co(OH)2. From the catalytic process of Co metal formed after the conversion reaction, the LiOH phase further reacts with lithium forming Li2O and LiH phases. With the contribution of LiOH, the theoretical capacity of Co(OH)2 is expanded by three times with 6 mol of lithium per unit formula [51].
As for the anode materials for SIBs, however, only a few studies have applied polyanionic compounds [92,97], and the additional redox reaction of sodium-containing species has not been observed.
4. Issues related to conversion-based anode materials
Despite the high reversible capacity compared to the insertion-based materials, many hurdles for the commercial usage of conversion-based materials remain. As shown in Table 1, there are large discrepancies in the capacity during the first discharge and charge. Furthermore, the large voltage hysteresis between the charge and discharge processes leads to a decrease in the energy efficiency. The decrease in capacity with repeated cycling owing to significant structural changes is another hindrance. These are critical issues for conversion-based anode materials that should be overcome.
4.1. Initial capacity loss (ICL)
The difference in capacity during the first discharge and charge is referred to initial capacity loss (ICL), as described in Fig. 4. For conversion-based anode materials, the ICL is particularly large compared to insertion-based materials, mainly caused by the following reasons: (i) formation of SEI layer with electrolyte decomposition, (ii) incomplete oxidation of metal, and (iii) irreversible side reaction of inactive species.
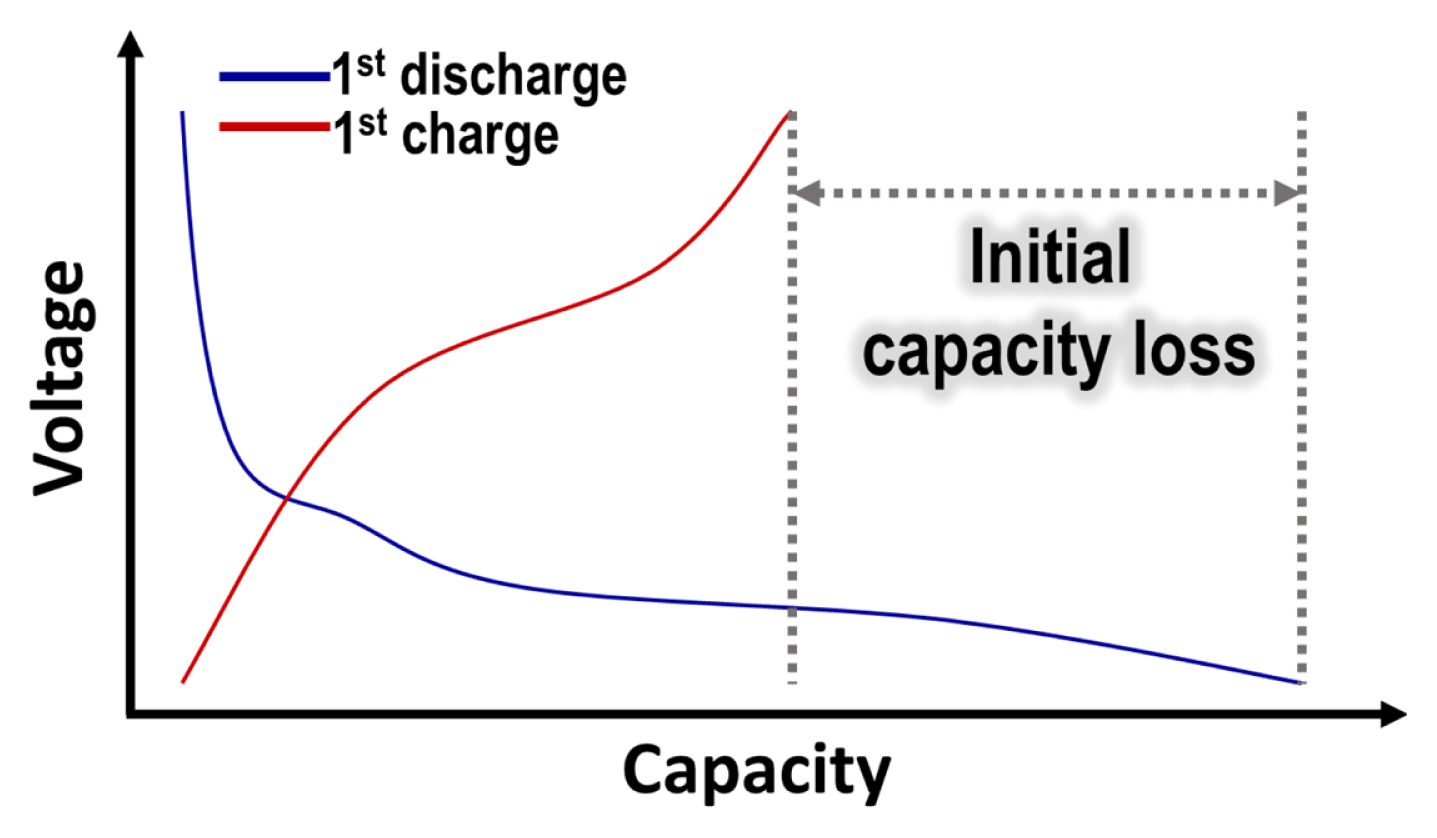
Typical voltage profiles of conversion-based anode materials during the first cycles, showing large ICL.
As mentioned in section 2.2, the surface layer is generated during discharge in conversion-based materials with a thickness of tens to hundreds of A. Previous reports have shown that these layers are composed of cation containing inorganic species (M2CO3, MF, M2O (M=Li, Na)) and organic species (hydrocarbons, polyethylene oxide, and alkyl carbonates) [40,41,128]. These results demonstrate that the surface layers are formed by decomposing electrolyte containing carbonates and solvated lithium/sodium salts. As the electrolyte solution is thermodynamically unstable at low potential, the electrolyte decomposition occurs at the surface of electrodes leading to the formation of a thick surface layer [129,130]. The electrolyte-derived surface layer can be decomposed partially reversibly, giving rise to reversible capacity, and the irreversible part protects the electrode surface from further decomposition [131]. However, a large number of cations are consumed during the formation of thick surface layer in the first cycle leading to a large ICL.
The conversion reaction was established based on the reactivity of Li2O. The decomposition of Li2O can occur in nanosized with the presence of catalytic metal species [13]. However, they cannot be decomposed alone because the conversion reaction products (Li2O, Na2O, etc.) are intrinsically inactive. As conversion-based materials undergo severe structural changes, especially during the first lithiation step, clustered metal atoms can lose electrical contact and be isolated by the surface layer. Thus, local deficiencies of metal nanoparticles are inevitable, hindering the complete decomposition of conversion reaction products [132]. Therefore, the oxidation state of the metal atoms after one complete cycle is generally lower than its original oxidation state before the electrochemical reaction [27,133,134]. These results indicate incomplete reversible conversion reaction, which is another cause of ICL.
Diverse irreversible side reactions from other components that are considered inactive also contribute to ICL. Some surface-adsorbed functional groups formed during material synthesis or electrode fabrication irreversibly react with the lithium [135,136]. As these functional groups are mainly generated at the surface, small active materials with large surface areas induce larger ICL than bulk ones. In addition, cation adsorption or trapping at the surface or defects inside active materials and even at conductive additives and binder also causes ICL [137,138].
4.2. Voltage hysteresis
As presented in Fig. 5, large potential difference during charge and discharge, called voltage hysteresis, is another major issue in conversion-based materials, significantly reducing energy efficiency during cycles. Compared to the insertion-based materials, the voltage hysteresis is much larger in conversion-based materials owing kinetic and thermodynamic reasons. The kinetically originated voltage hysteresis is ascribed to the slow mobility of the charge carrier and the accompanying redistribution of metal atoms that occur during the conversion reaction [139,140]. In conversion-based anode materials, cation diffusion is restricted by the interfacial resistance between the two phases and the thick surface layer. While the kinetic voltage hysteresis is highly dependent on the applied current, the other voltage hysteresis group from thermodynamical reason is irrelevant and rather intrinsic [141,142]. The thermodynamic voltage hysteresis is attributed to different reaction pathways between the ion storage and release process, and increase in free energy by increased surface area and formation of amorphous state after the conversion reaction [11].
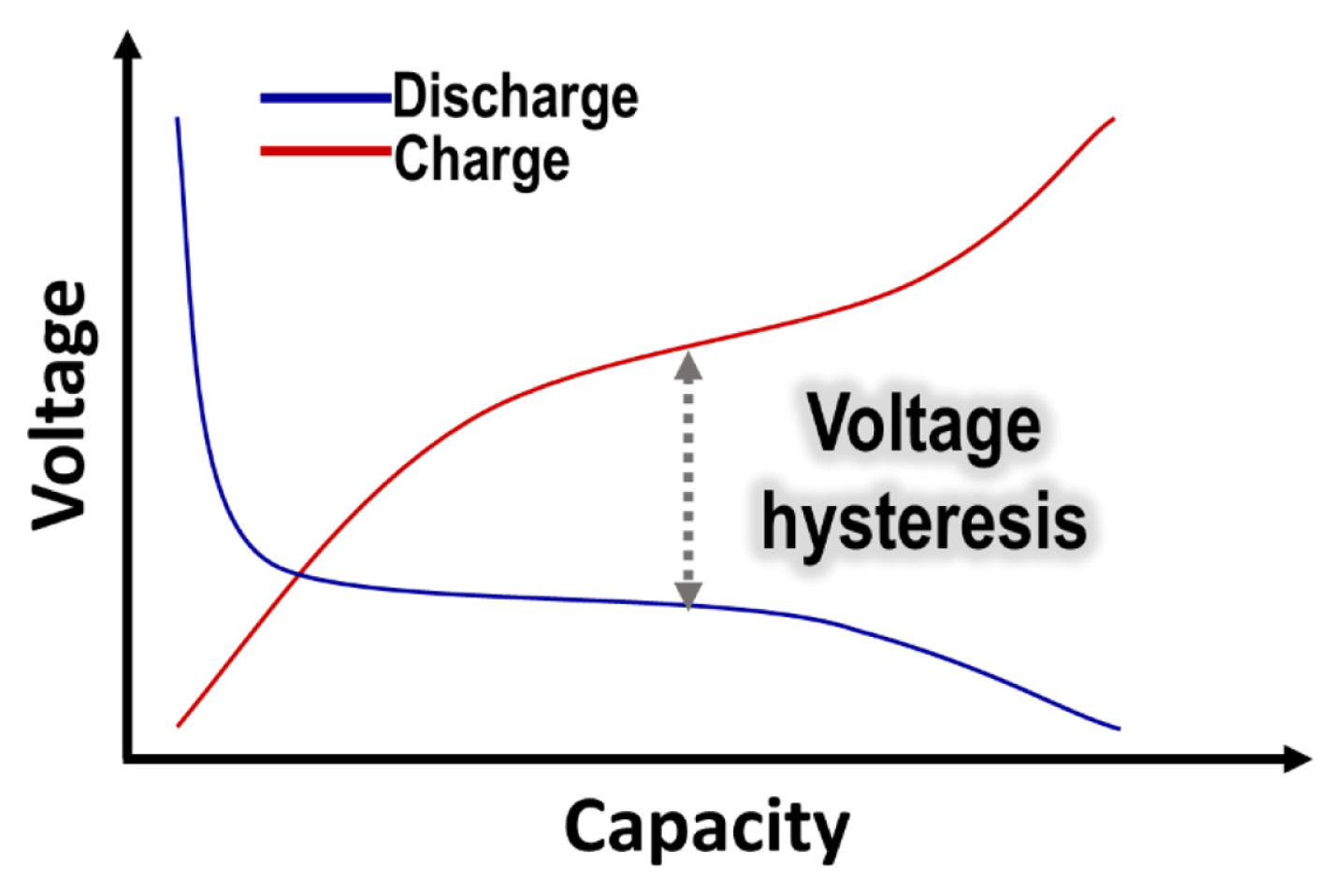
Typical voltage profiles of conversion-based anode materials after the first cycle, showing voltage hysteresis.
H. Kim et al. scrutinized the origin of the voltage hysteresis of conversion-based Co3O4 anode materials for SIBs. Using XAS, XPS, and TEM, the authors show that thermodynamic voltage hysteresis is the main contributor, owing to the different reaction pathways during the discharge and charge processes [143]. During sodium-ion insertion, Co3O4 is directly converted into metallic Co and Li2O. In contrast, metallic Co was first converted to the intermediate CoO1−x phase and further oxidized to nanosized Co3O4 during charge. The asymmetric reaction pathway in conversion-based materials can also be observed in other conversion-based anode materials in LIBs and SIBs [31,144,145]. Considering kinetics, the slow diffusion kinetics of metal or ligand in the host structure during conversion reaction also increase the voltage hysteresis [44].
4.3. Capacity fading
As large amount of lithium- and sodium-ions can be stored in conversion-based anode materials, considerable volume expansion and shrinkage up to 200–300% occurs during discharge/charge process as evidently observed by in situ TEM analysis [21,35]. The repeated volume changes during electrochemical cycling lead to particle pulverization and electrical contact loss of active particles [15,146]. In addition, nanosized particles can be agglomerated and further undergo this process [147]. These severe morphological changes result in rapid capacity fading and limited cycle life, as displayed in Fig. 6.
Typical relation between cycle number vs. specific capacity of conversion-based anode materials.
Additionally, continuous growth of the surface layer consumes the available cation sources and hinders the diffusion of charge carriers. J. Li et al. inspected the reason for the capacity fading of Fe3O4 by observing the phase evolution process using synchrotron-based X-ray techniques and in situ TEM analysis [27]. As the electrochemical cycling proceeds, the surface layer continuously grows on the active particle by accumulation of Li2O due to an incomplete reversible conversion reaction and electrolyte decomposition. The thick surface layer leads to a significant increase in diffusion length, acting as a kinetical barrier against electron transport. Thus, the authors insist that the growth of the surface passivation layer is the main factor for capacity fading, which also results in poor rate capability.
Conversely, changes in the surface layer occasionally cause beneficial anomalous phenomena of capacity increase during repeated cycling, known as negative fading [46,148,149]. H. Sun et al. inspected the morphological changes in mesoporous Co3O4 during cycling under high specific current [150]. During the initial 90 cycles, the reversible capacity sharply decreased from ~830 mAh g−1 to ~ 250 mAh g−1 due to the growth of the surface layer, cracking and pore coalescence of hollow particles, which can be generally observed in conversion-based anode materials. Interestingly, the reversible capacity gradually increased to ~900 mAh g−1 during the subsequent cycle, and then stabilized. The increase in the capacity originates from the changes in morphology. After 850 cycles, the thickness of the surface layer decreased, and the pores expanded, enhancing the kinetic properties again.
5. Strategies for improving the performance of conversion-based anode materials
To establish high-capacity conversion-based anode materials for commercial use, the above-mentioned critical disadvantages should be addressed. To date, numerous research efforts have been devoted to providing clues to overcome these issues and improve electrochemical performance. Some useful strategies are categorized as nanoscale engineering, surface coating and artificial SEI layer, polymorph control, and introducing extra catalytic elements. These strategies improve the kinetic properties, enhance the reversibility of the conversion reaction, or inhibit side reactions, all of which positively affect electrochemical performance.
5.1. Nanoscale Engineering
Synthesizing nanosized materials with specially designed morphologies is the most widely used strategy. Nanoscale engineering effectively reduces the diffusion length and provides a void space for buffer volume expansion. Thus, nanostructured materials generally exhibit better rate and cycle performance than micron-sized materials [151–153]. Moreover, nanomaterials with unique morphologies such as hollow, mesoporous, and hierarchical structures exhibit better performance than simple nanoparticles [15,154,155]. L. Yu et al. synthesized hollow prisms composed of nanosized CoS2 bubble-like subunits as shown in Fig. 7(a)–(b) [156]. The electrochemical performance of the resultant CoS2 nanobubble hollow prisms is provided in Fig. 7(c)–(e). It exhibits a high reversible specific capacity of ~861 mAh g−1 at 200 mA g−1 current. At an extremely high specific current of 5 A g−1, CoS2 hollow particles stably operate with a specific capacity of ~470 mAh g−1, and the capacity recovers to ~864 mAh g−1 when the current density is reduced to 200 mA g−1 again. After 200 cycles at 200 mA g−1, high reversible capacity of ~737 mAh g−1 was achieved, which is ~85% of the capacity at the second cycle. The excellent rate and cycle performance of CoS2 are attributed to the unique structural features. The kinetic properties are improved owing to the short lithium-ion diffusion pathway, and the interior void spaces alleviate the structural stress during repeated volume changes.

(a) SEM and (b) TEM image of CoS2 nanobubble hollow prisms. (c) Voltage profiles of CoS2 hollow prisms at 200 mA g−1. (d) Rate and (e) cycling performance of the CoS2 hollow prisms. (a)–(e) Reproduced with permission from Ref. [156] Copyright © 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (f) Changes in Sn-O bond strength during the conversion reaction (left upper), Sn K-edge XANES spectra during the deconversion reaction (right upper), and voltage profiles of SnO2/rGO composite electrode. Reproduced with permission from Ref. [69] Copyright © 2019 Elsevier B.V.
Compositing nanosized active particles with conductive carbonaceous materials such as graphene and carbon nanotubes is also an effective solution for improving electrochemical performance. Carbonaceous materials prevent the agglomeration of nanoparticles and provide an additional electron diffusion pathway. Thus, the cyclability and rate capability of carbon composite materials are greater than those of bare materials because of the enhanced kinetic properties and stable carbonaceous matrix during ion repeated storage/extraction cycles [157–161]. In addition to the cycle and rate performance, carbon composite materials generally exhibit higher capacity than pure active materials or pure carbonaceous materials [39,69,162]. This improvement in capacity is called a synergistic effect, originating from the intimate contact between carbon and active materials. The intimate contact prevents particle aggregation of nanoparticles during synthesis and repeated cycling, and enhances both ion and electron diffusivity [163,164]. Furthermore, carbonaceous materials affect the intrinsic physicochemical properties of the active materials, enhancing the reversibility of the conversion reaction [39,69]. As shown in Fig. 7(f), H. Kim et al. show that the oxidation state of SnO2/graphene composite notably increased while bare SnO2 shows only a slight change during charging over 1.5 V where the reversible alloying reaction of Sn is completed. This indicates that the reversibility of the conversion reaction is much higher in the composite material. The improved reversibility is attributed to the weakened Sn-O bond strength; thus, less energy is required to break and form bonding between Sn and O atoms [69].
5.2. Surface coating
Although nanoscale engineering in the fabrication of active materials is beneficial for electrochemical performance in terms of cyclability, rate capability, and reversible capacity, the increased surface area could result in a high ICL due to a higher chance of unexpected side reactions. To reduce side reactions, coating stable layers on the surface of active particles is helpful. These surface protection layers can prevent electrolyte decomposition, suppress the growth of thick SEI layers, and buffer volume expansion, leading to higher efficiency and longer cycle life expectancy. However, the coating layer should be extremely thin, as the inactive surface layer could also hinder ion/electron transport if the coating layer is thick.
Atomic layer deposition (ALD) is a powerful technique for achieving uniform thin coating on the surface of particles. The ALD coating process involves sequential alternating pulses of different gaseous precursors. First, one precursor is pulsed and reacts with the functionalized substrate to uniformly cover the surface. Another precursor pulse is then inserted into the chamber, which reacts with the first precursor. After each step, the chambers are purged with an inert gas to remove the remaining unreacted precursors. These steps, called one ALD cycle, create one coating layer with desired materials. By repeating the ALD cycles, the thickness of coating layers can be controlled at the angstrom level [165]. Some oxides such as Al2O3 [166–168], TiO2 [169], and HfO2 [170,171] are widely applied as coating materials using ALD.
B. Ahmed et al. uniformly coated MoO3 nanorods with HfO2 by ALD [172]. As shown in Fig. 8(a)–(d), the thickness of the HfO2 coating layer is uniform and directly proportional to the number of ALD cycles. As anode materials for LIBs, HfO2-coated MoO3 exhibits greater electrochemical performance than the uncoated materials, as shown in Fig. 8(e)–(f). When cycling under a high specific current of 1 and 1.5 A g−1, the specific capacity of bare MoO3 gradually decreases, while HfO2-coated MoO3 delivers stable capacity. Although HfO2 is an inactive passivation layer, the coated materials exhibit better rate capability. At a higher specific current of 1.5 A g−1, the coated materials still exhibit a higher capacity of ~742 mAh g−1 than ~534 mAh g−1 in bare MoO3. According to the authors, the uniform thin HfO2 coating does not critically block the lithium-ion diffusion and prevent structural degradation, leading to performance improvement.
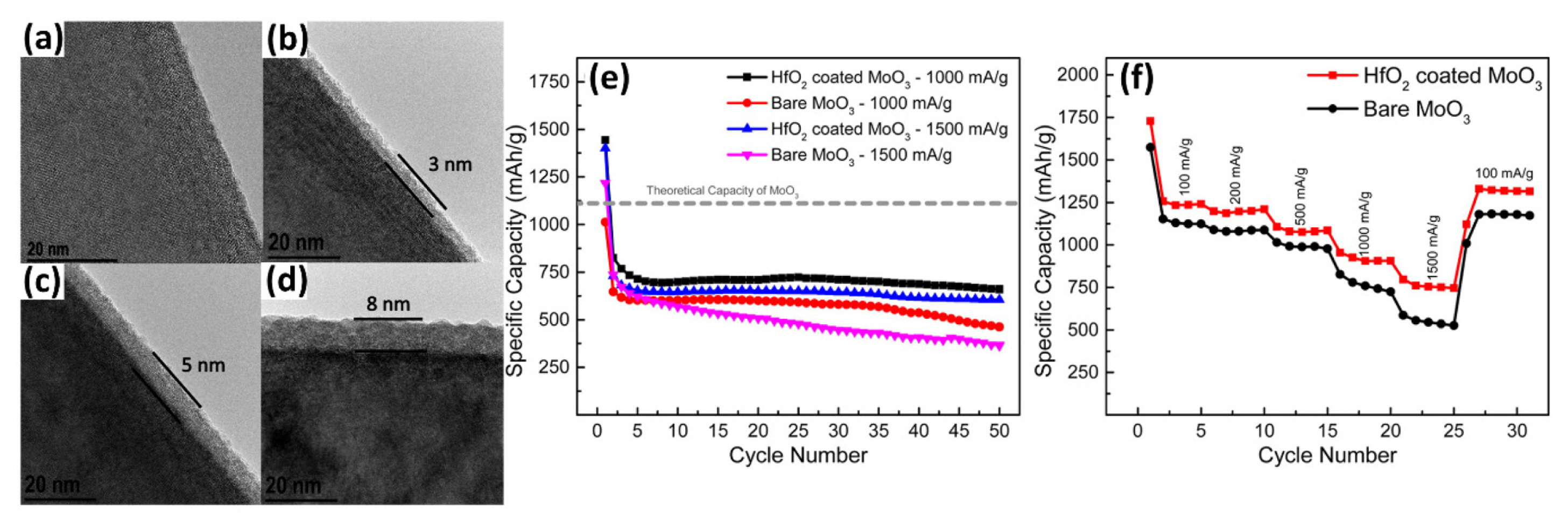
High-resolution TEM images of (a) bare MoO3 and HfO2-coated MoO3 powders after (b) 30, (c) 50, and (d) 80 ALD cycles. (e) Cyclic performance at 1000 and 1500 mA g−1 and (f) rate capability of bare MoO3 and 10 ALD HfO2-coated MoO3. Reproduced with permission from Ref. [172] Copyright © 2015 American Chemical Society.
Additionally, uniform coating of carbonaceous materials improves the surface stability of conversion-based anode materials [173–175]. X. Li et al. coated SnS nanoflakes with carbon (SnS@C) via chemical vapor deposition (CVD) using ethanol as carbon sources [173]. The resultant SnS@C exhibits ~810 and ~613 mAh g−1 as anode materials for SIBs under a specific current of 0.2 A g−1 during the first discharge and charge processes, respectively. The coulombic efficiency in the first cycle of SnS@C is ~75.7%, which is higher than that of bare SnS. In addition, SnS@C exhibits better rate and cycle performance than bare SnS anode materials, as the surface carbon coating layers not only endure the volume changes but also provide a conductive matrix.
5.3. Electrolyte additives
Although the irreversible capacity is the largest at the first cycle, the coulombic efficiency of conversion-based materials at subsequent cycling is also generally below 100% because the indecomposable surface layer derived from electrolytes continues to grow even after the first formation cycle. The growth of the surface layer further hinders lithium-ion transport and consumes active lithium sources, leading to rapid capacity fading. Incorporating a small amount of foreign electrolyte additive is a cost-efficient approach to prevent electrolyte decomposition after the initial formation cycle. The electrolyte additives function as sacrificial components to be preferentially decomposed prior to the main electrolyte species, forming a more stable surface passivation layer. Thus, electrolyte additive should have a lower lowest unoccupied molecular orbital (LUMO) than the main electrolyte species [176,177]. Vinylene carbonate (VC) and fluoroethylene carbonate (FEC) are frequently used as electrolytes. These carbonate-based additives contribute to forming flexible long polycarbonate chains, resulting in the formation of a stable SEI layer [178,179]. In addition, the fluorine-containing additive leads to the formation of LiF species on the surface, which efficiently prevents the growth of the surface layer [180,181]. Therefore, the coulombic efficiency and cycle performance of conversion-based materials can be enhanced from these electrolyte additives [180,182,183].
Z. Zhu et al. investigated the beneficial effect of FEC additive in conversion-based MoS2 electrode materials, as provided in Fig. 9 [180]. Without FEC additive, the surface layer on the MoS2 electrode becomes thicker and denser with cycling. Therefore, the charge transfer resistance increases from 25 Ω at the 25th cycle to 120 Ω at the 100th cycle. However, after adding FEC, the surface morphology and the surface layer thickness do not change significantly during repeated cycling. In addition, the resistivity increases only slightly, from 25 Ω at the 25th cycle to 32 Ω at the 100th cycle. The FEC additive is reduced prior to the main electrolyte solvent generating a stable and thin LiF-rich surface layer on the MoS2 electrode, which endures volume changes and prevents continuous electrolyte decomposition during repeated cycling. As a result, the MoS2 anode material stably exhibits high capacity of ~770 mAh g−1 during 200 cycles at specific current of 1 A g−1, whereas capacity rapidly decreases after 50 cycles in FEC-free electrolyte.
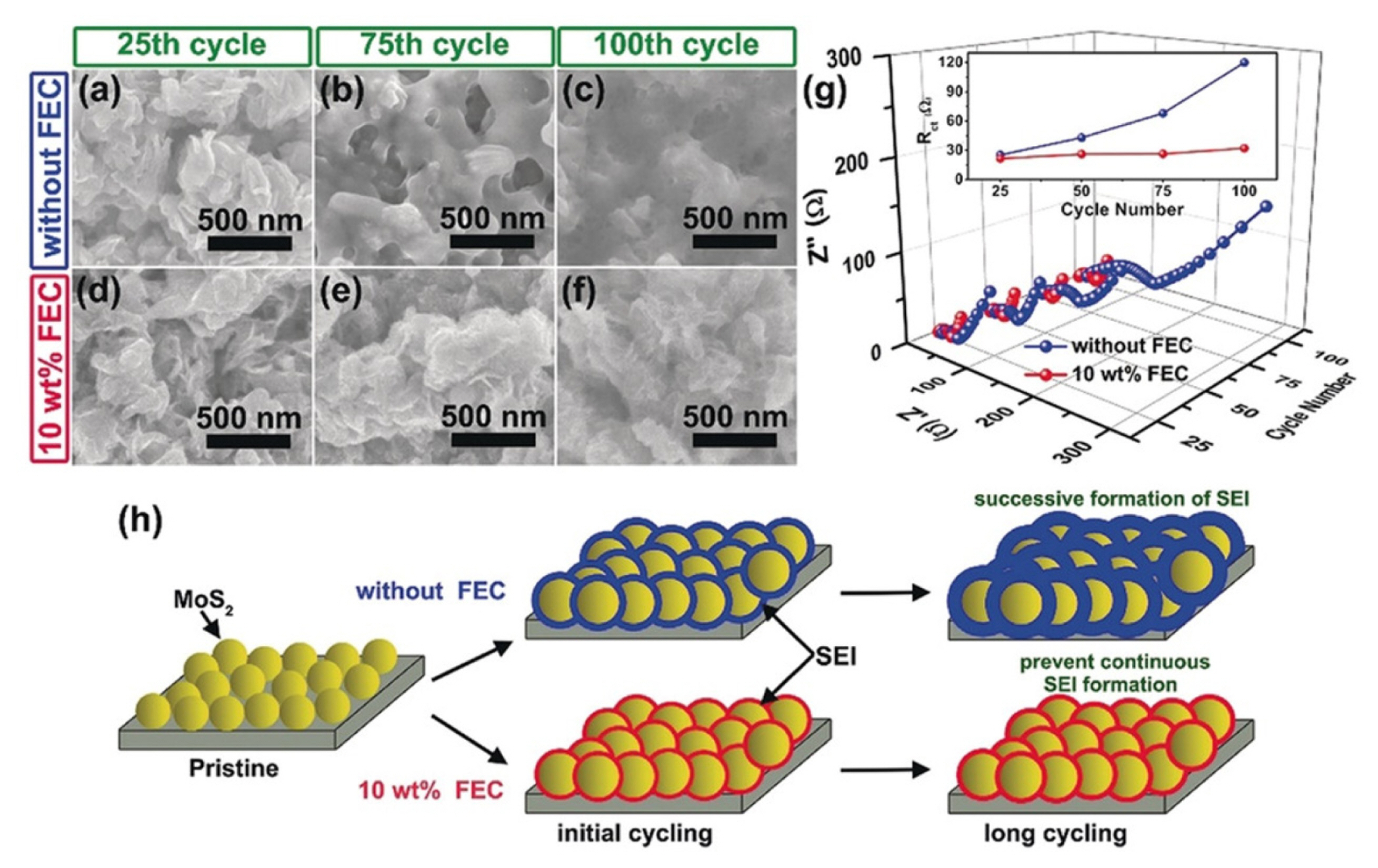
Surface morphology of MoS2 electrode after different cycles (a)–(c) without FEC and (d)–(f) with FEC additive. (g) electrochemical impedance spectroscopy result after cycling at different electrolyte conditions. (h) Schematic illustration of surface layer formation on the MoS2 electrode without and with 10 wt% FEC additive. Reproduced with permission from Ref. [180]. Copyright © 2018 Wiley-VCH GmbH.
5.4. Polymorph control
In conversion-based anode materials, the crystal framework collapses into nanosized metal and loses crystallinity. Therefore, the choice of polymorphs was not considered to be significant compared to the other factors. However, a few reports have shown that the electrochemical performance varies with the polymorphs [184–187]. K. Chen et al. prepared α-, β-, and γ-MnO2 using microwave-hydrothermal process and compared their electrochemical performance as anode materials for LIBs [188]. Among these materials, γ-MnO2 exhibits the highest reversible capacity ~600 mAh g−1. They attributed the high performance of γ-phase to the suitable tunnel size (1 × 2) for lithium-ion diffusion. Contrastingly, the diffusion is hindered by the compact tunnel size (1 × 1) in β-MnO2 and potassium ions located in the (2 × 2) tunnel in α-MnO2. However, the morphologies of the three samples were different, and the reversible capacity was far below the theoretical capacity; thus, it cannot represent the effects of polymorphs in conversion-based MnO2 anode materials.
To explore the effects of polymorphs, H. Kim et al. systematically investigated the charge storage behaviors of four different MnO2 polymorphs possessing similar hollow porous morphology, as shown in Fig. 10(a) [24]. The voltage profiles of the four samples in Fig. 10(b) are similar except for the relatively high potential region over ~1.0 V during the initial discharge, where the insertion reaction occurs. Using synchrotron-based analyses, they observed that the charge storage reactions are identical. During discharge, MnO2 is sequentially converted to MnO and Mn metal/Li2O. After delithiation, interestingly, spinel-phased λ-MnO2 is formed in four samples instead of returning to the original polymorphs. Despite the identical reaction mechanism, λ-MnO2 delivers the highest reversible capacity of ~1270 mAh g−1 among the four MnO2. To investigate the origin of different electrochemical reactivity, the kinetic properties were compared using the galvanostatic intermittent titration technique (GITT) as depicted in Fig. 10(c). The GITT results indicate that the overpotential is the smallest in λ-MnO2 owing to the structural similarity between the initial and cycled states, which results in the high reversibility of the conversion reaction.

S. Hariharan et al. reported the lithium storage property of α-and γ-Fe2O3 [189]. Both Fe2O3 transformed to metallic Fe and Li2O phases during the initial lithiation, and after full charge, nanosized γ-Fe2O3 was observed in both materials. Such transition to γ-Fe2O3 is attributed to different thermodynamics at nanoscale. Although α-Fe2O3 is the stable phase at bulk scale, γ-Fe2O3 is more stable in nanosized materials. In addition, the authors insisted that the excess energy generated during the transformation of the initial micron-sized materials to nanoscale γ-Fe2O3 is larger in γ-Fe2O3 electrode, leading to relatively well-crystallized particles after the electrochemical cycle. The well-crystallized products and structural similarity between the initial and cycled states in γ-Fe2O3 electrode result in enhanced rate performance during the electrochemical conversion reaction.
These results show that the polymorphs also affect electrochemical performance and should be considered when designing conversion-based electrode materials. It is beneficial to choose the most thermodynamically stable polymorphs after the electrochemical cycle and possess advantageous intrinsic properties. L. Zhang et al. utilized tetragonal-FeSe (t-FeSe) with metallic conductivity as a conductive additive-free electrode material for SIBs and LIBs [187]. In situ XRD experiments during sodium and lithium storage/extraction cycle show that t-FeSe is reversibly formed instead of transformation to the hexagonal phase, maintaining the structural similarity with the pristine state. Compared with the hexagonal FeSe exhibiting only a small capacity below ~150 mAh g−1, t-FeSe shows superior electrochemical sodium-ion storage performance, delivering a capacity of ~400 mAh g−1 at a rate of 0.5 C for 30 cycles. Even at a faster rate (12.5 C), relatively high capacity of ~280 mAh g−1 was exhibited. In addition, owing to the absence of conductive additive, the tetragonal FeSe electrode can achieve remarkably high energy density for both SIBs and LIBs.
5.5. Introducing extra catalytic elements
The reversible conversion reaction involves the decomposition of LiX or NaX phases that were considered inactive, with the aid of metal nanoparticles formed after the conversion reaction. Therefore, the reversibility of the conversion reaction is correlated with the catalytic properties of metal nanoparticles. Introducing heterogenous metal elements, which act as catalysts, is an effective strategy for enhancing the reversibility of the conversion reaction.
In conversion-based anode materials, the oxidation state of metal after one complete cycle is generally lower than that of the pristine state, as the conversion reaction is not fully reversible. The addition of heteroatoms is advantageous for improving the reversibility of conversion reaction. For example, doping Co, Ni, and Zn in manganese compounds enables the oxidation of Mn2+ to a higher oxidation state during charging [190,191], which is kinetically restricted under normal conditions. Likewise, Co doping has been reported to increase reversible capacity in NiO, MoO3, and MoS2 anode materials, by the catalytic activity of additional Co sources [192–194].
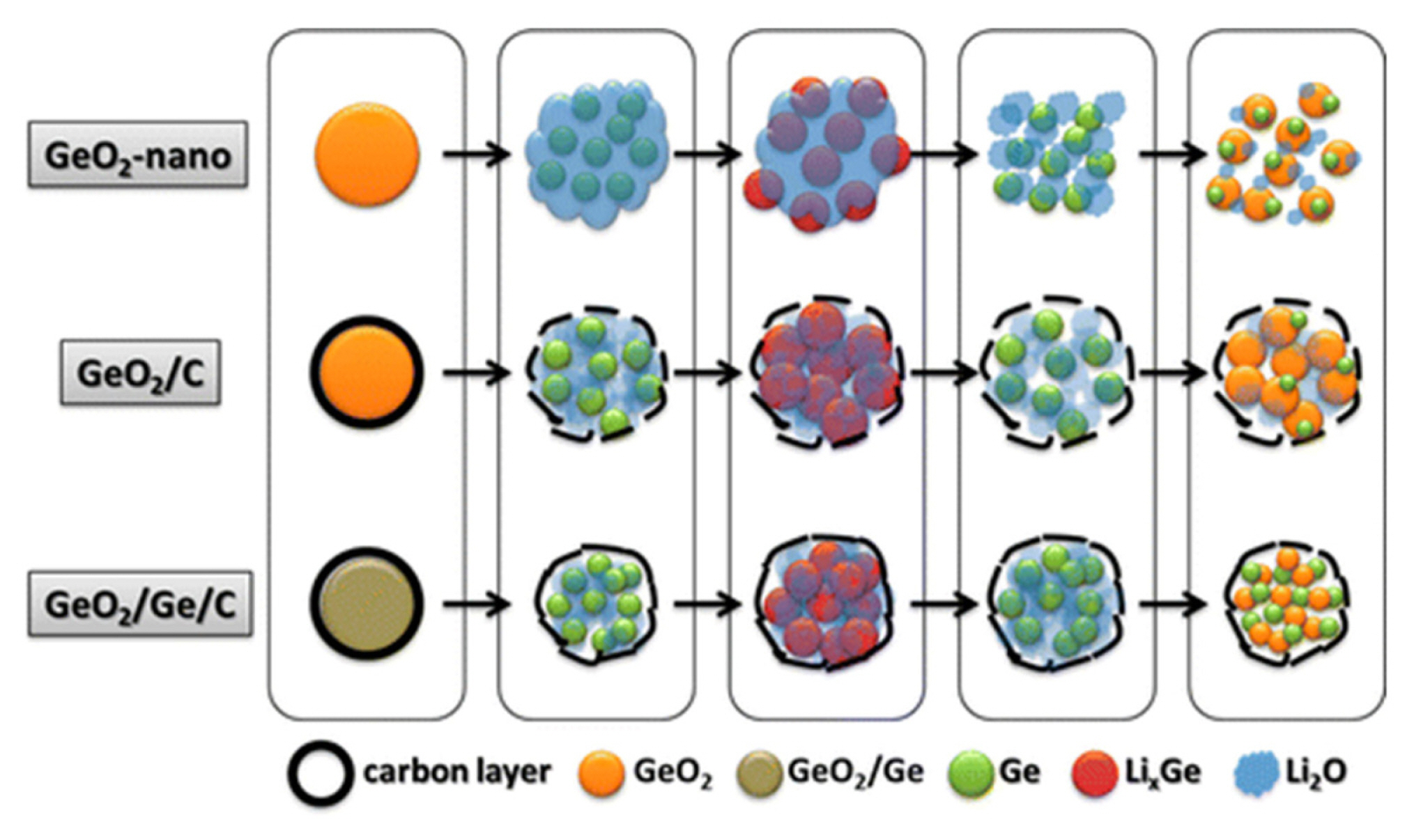
Metal compounds containing lithium and sodium alloying metals (MX, M= Zn, Sn, Ge; X=O, S, Se) commonly exhibit large irreversible capacity due to a limited reversible conversion reaction. In these compounds, sequential conversion and alloying reactions occur during the first lithiation step. At the following charge, however, only the dealloying reaction occurs almost completely, while the conversion reaction is only partially reversible [29,195–197]. To reduce the irreversible capacity loss in these materials, various metal species, such as Fe, Co, Ni, Cu, and Ge, were added to the compounds to facilitate the reoxidation of metal nanoparticles [198–203]. The addition of alien metal species improves electronic and ionic connections and exerts catalytic effects for decomposing LiX and NaX phases, enhancing the reversibility of the conversion reaction. In addition, the metal atoms that do not form alloys with lithium or sodium alleviate the volumetric strain, improving the cycle performance. For instance, additional germanium in GeO2/C composite (Ge/GeO2/C) increases the reversible capacity to ~1800 mAh g−1 at 1 C rate with relatively high initial coulombic efficiency, while bare GeO2/C exhibits ~1400 mAh g−1 [203]. The superior electrochemical performance originates from the active decomposition of Li2O by the catalytic effect of the elemental germanium, as schematically described in Fig. 11.
5.6. Utilizing abnormal charge storage reaction
As briefly mentioned in chapter 2, numerous reports have shown that the actual reversible capacity of conversion-based materials surpasses the theoretical capacity owing to the contribution of abnormal charge storage reactions. If the additional capacity can be controlled and fully utilized, the energy density of electrode materials would be maximized, leading to a revolutionary development in the field of energy storage. Therefore, the origins of additional capacity have attracted much attention [46,204,205], starting from the early 2000s [48,49]. The most widely accepted reasons for the additional capacity of conversion-based materials are electrolyte-derived surface layer [48], interfacial charge storage [49,50], and reactions of lithium-containing species [43,51,90].
As these reactions are considerably affected by the morphological features and catalytic properties of active materials, nanoscale engineering and the introduction of extra catalytic elements facilitate abnormal charge storage reactions. By reducing the size of active materials, the specific surface area is enlarged, causing more surface reactions with electrolytes. Therefore, capacity from the reversible formation/decomposition of electrolyte-derived surface layers increased in nanosized electrodes [151,206]. The nanomaterials generate more interfacial area between the metal phases and LiX/NaX, and thus, a larger amount of cation can be stored at the interface [68]. Additionally, electron-conducting carbonaceous materials can act as electron reservoirs when in contact with ionic conducting phases, similar to metal phases, exhibiting additional capacity by interfacial reactions [207–209].
As mentioned in section 3.4, additional lithium storage reactions of conversion reaction products occur in polyanionic metal compounds. These reactions of lithium-containing species formed after conversion reaction are also enabled by the catalytic effects of nanosized metal. Therefore, mixing metal species to obtain optimal catalytic property improves electrochemical performance compared to single metal compounds [210–212].
J. H. Um et al. synthesized high-performance tindoped ferrite composited with reduced graphene oxide (Fe2.76Sn0.24O4/rGO) [134]. Fig. 12(a) shows that the negative fading can also be observed in Fe2.76Sn0.24O4/rGO as in mesoporous Co3O4 [150], but it is originated from different reasons. As shown in Fig. 12(b), the oxidation state of Fe is higher after the 100th cycle than after the 1st cycle. After the first cycle, the Fe oxidation state is correlated with the Sn content, because Sn catalyzes the decomposition of Li2O, leading to increased reversibility of the conversion reaction as in chapter 5.4. After 100 cycles, the oxidation state is even higher than that of the pristine state owing to the formation of the FeOOH phase, confirmed by EXAFS as shown in Fig. 12(c). The FeOOH phase is generated by the reaction between the Fe metal and LiOH in the surface layer, implying an enhanced contribution of the reversible reaction of the electrolyte-derived surface layer. They insist that the increased contribution of the surface layer reaction and complete reoxidation are the reasons for the negative fading. Therefore, the catalytic material design could also facilitate the reaction of electrolyte-derived surface layer, improving electrochemical performance.

(a) Cycle performance of all the Fe3-xSnxO4/rGO composites and bare Fe2.76Sn0.24O4 without rGO. (b) Comparison of the Fe oxidation states in Fe3-xSnxO4/rGO composites. (c) Fourier transformed EXAFS spectra of Fe2.76Sn0.24O4/rGO composite in the pristine state and after the 1st and 100th cycled states. Reproduced with permission from Ref. [134] Copyright © 2019 American Chemical Society.
6. Conclusions
This review presents a keen understanding of the conversion-based materials for LIBs and SIBs. Conversion reaction occurs with the formation of metal nanoparticles embedded in amorphous LiX/NaX phases. During lithiation, the volume significantly expands, and thick surface layers are formed. At subsequent charging, the surface layer and LiX/NaX are reversibly decomposed by the catalytic activity of metal nanoparticles. As these cycling processes involve a large number of mobile ions, the reversible capacity of conversion-based materials is much higher than that of insertion-based materials, often above ~1000 mAh g−1. However, several issues of ICL, voltage hysteresis, and capacity fading have to be overcome for commercial use. The ICL originated from (i) irreversible formation of indecomposable surface layer, (ii) limited reversibility of conversion reaction, and (iii) unexpected side reactions of inactive species. Voltage hysteresis occurs by asymmetrical reaction pathways between charging and discharging, and slow reaction kinetics. The capacity fading originated from repeated volume changes, leading to detrimental morphological changes and continuous growth of passivation layers. Several materials design strategies have been developed to get through these issues, potentially further improving the electrochemical performance of conversion-based materials.
Acknowledgment
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2019R1A2C2003731).