1. Introduction
Since the commercialization of LIBs in 1991 and in line with the rapid market growth of electric vehicles and energy storage systems, the demand for high-energy-density batteries has steadily increased [1–3]. However, commercialized graphite can only theoretically deliver a low capacity of 372 mAh g−1 [4], which cannot satisfy market expectations requiring higher energy and power densities. Accordingly, novel anode materials such as metal oxides [5,6], metal hydroxides [7,8], and silicon-based anodes [9,10] are being investigated as candidates for next-generation anode materials. These materials show a significantly high energy density than graphite. However, many drawbacks still exist, such as extreme volume changes during cycling and poor reaction kinetics [11,12]. To overcome these issues, compositing these materials with carbonaceous materials has been actively studied, which exhibit more excellent electrochemical performance [13–15].
Among these candidates, Co(OH)2 has been widely studied as an intriguing candidate for anode materials owing to its high specific capacity, which is beyond the theoretical value based on the conventional conversion reaction mechanism [8,16,17]. It was reported that the extra capacity originated from an additional reaction of LiOH in the low-voltage region in the presence of nanosized Co metal [17,18]. Furthermore, graphene-composited Co(OH)2-based anode materials exhibit significantly larger capacities than bare materials [8,15,19]. Although the reaction mechanism of Co(OH)2 has been scrutinized as the conversion reaction and the reaction of LiOH [17], it remains unclear how the reversible capacity improves after compositing Co(OH)2 with graphene.
In this study, a Co(OH)2/graphene nanoplatelet (GNP) composite was synthesized to compare the electrochemical properties and charge-storage reactions of Co(OH)2/GNP and bare Co(OH)2. The synthesized composite materials showed a high reversible capacity of ~1090 mAh g−1, which is 1.8 times the theoretical value and much larger than the reversible capacity of bare Co(OH)2. To investigate the exact lithium storage mechanisms and the reason for these improvements, combined analyses of X-ray absorption spectroscopy (XAS) and electrochemical characterizations were performed. Systematic analyses demonstrated that both conversion and LiOH reactions were facilitated in the Co(OH)2/GNP composite. We believe that in addition to providing insights into the design of electrode materials with graphene composites, our findings could improve current understanding of the exceptional lithium storage mechanism.
2. Experimental
2.1. Material preparation
Co(OH)2/GNP powder was prepared by using cobalt (II) hydroxide (bare Co(OH)2, Sigma-Aldrich) and graphene nanoplatelet aggregates (GNP, S.A. 500 m2 g−1, Alfa Aesar) as precursors. The synthesis procedure followed a simple one-step process proposed in a previous report [20]. First, 0.2 g of Co(OH)2 and 0.025 g of GNP were dispersed in 50 mL of deionized (DI) water by sonication for 2 h to exfoliate the graphene sheets and Co(OH)2 layers. Subsequently, the solution was filtered several times with DI water to restack graphene and Co(OH)2 layers and to remove impurities. The resultant precipitates were then dried at 70°C for 24 h in a vacuum oven.
2.2. Material characterization
Laboratory-scale X-ray diffraction (XRD) patterns of the synthesized materials were obtained using a D2 PHASER diffractometer (Bruker Corp.) in the range of 10° < 2θ < 20° with Cu Ka radiation (λ = 1.5406 Å). To estimate the graphene content in Co(OH)2/GNP, thermogravimetric analysis (TGA) was conducted using a SEIKO INST (TG/DTA 7300) at a temperature range of 25–800°C in air flow. The morphology was observed using high-resolution transmission electron microscopy (TEM, JEOL-JEM-2100F). X-ray absorption spectra (XAS) were obtained at the 8C beamline of the Pohang Light Source-II (PLS-II) in transmission mode with ionization chambers at room temperature. The Co K-edge spectra of the samples were obtained simultaneously with the reference spectra using a Co metal foil for energy calibration. XAS data processing, including background subtraction, normalization, k3-weighting, and Fourier transform, was conducted using the Athena software to obtain X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure spectroscopy (EXAFS) spectra [21]. For ex situ XAS analysis, electrodes were collected by disassembling the coin cells in an argon-filled glove box after reaching the specific states of charge and then washed with diethyl carbonate solvent.
2.3. Electrode preparation and electrochemical analysis
To prepare the working electrodes, a slurry with a weight ratio of 7:1:2 of the active materials, conductive additive (Ketjenblack, EC-300J), and binder (polyamide-imide (Solvay) dissolved in N-methyl pyrrolidone (Aldrich)) was mixed in a mortar. The mixed slurry was uniformly coated on a Cu foil and subsequently dried at 200°C for 3 h under vacuum. The fabricated electrodes were assembled into CR2032 coin cells with metallic lithium foil as the counter electrode and Celgard separator. Subsequently, 1.3 M of LiPF6 dissolved in a solvent consisting of a mixture of ethylene carbonate/diethyl carbonate (v/v = 3:7; Dongwha electrolyte) was used as the electrolyte. Galvanostatic charge/discharge measurements were performed using a WonATech battery cycler (WBSC3000S). The cut-off voltage range was set as 0.001–3.0 V or 0.001–1.5 V with a constant specific current of 100 mA g−1 for the cycling test. The galvanostatic intermittent titration technique (GITT) was conducted using the same machine by repeatedly applying a constant current for 30 min and resting for 3 h. Cyclic voltammetry (CV) was conducted at a scan rate of 0.1 mV s−1 in the voltage range of 0.001–3.0 V.
3. Results and Discussion
As shown in Fig. 1(a), the XRD pattern of the Co(OH)2/GNP composite is similar to that of commercial bare Co(OH)2 and adequately matches the JCPDS reference spectra (PDF card No. 30-0443) without any impurities. The oxidation state of Co and the local structures were investigated using Co Kedge XAS experiments. The Co K-edge XANES spectra in Fig. 1(b) demonstrate that the absorption edge positions of the bare Co(OH)2 and Co(OH)2/GNP composites are close to that of commercial CoO, demonstrating that the oxidation state of Co is 2+. The Fourier-transformed EXAFS spectra of the two materials in Fig. 1(c) are analogous, which indicates that the local structures around the Co atoms in Co(OH)2/GNP and bare Co(OH)2 are identical. These results demonstrate that the Co(OH)2 phase was well-maintained during the synthesis of the composite materials. TGA experiments were conducted to investigate the GNP content in the composite materials. As shown in Fig. 1(d), the weight loss during heating to 800°C was approximately 16.5% and 27.8% for bare Co(OH)2 and Co(OH)2/GNP, respectively. Therefore, the GNP accounts for the 11.3% of Co(OH)2/GNP, which is removed at over 300°C. The remaining weight loss was due to the evaporation of surface-absorbed water and dehydration during the transformation of Co(OH)2 to Co3O4 [22,23]. TEM also confirmed the presence of GNPs in the Co(OH)2/GNP composite materials, as shown by the amorphous particles between the hexagonal Co(OH)2 layers.
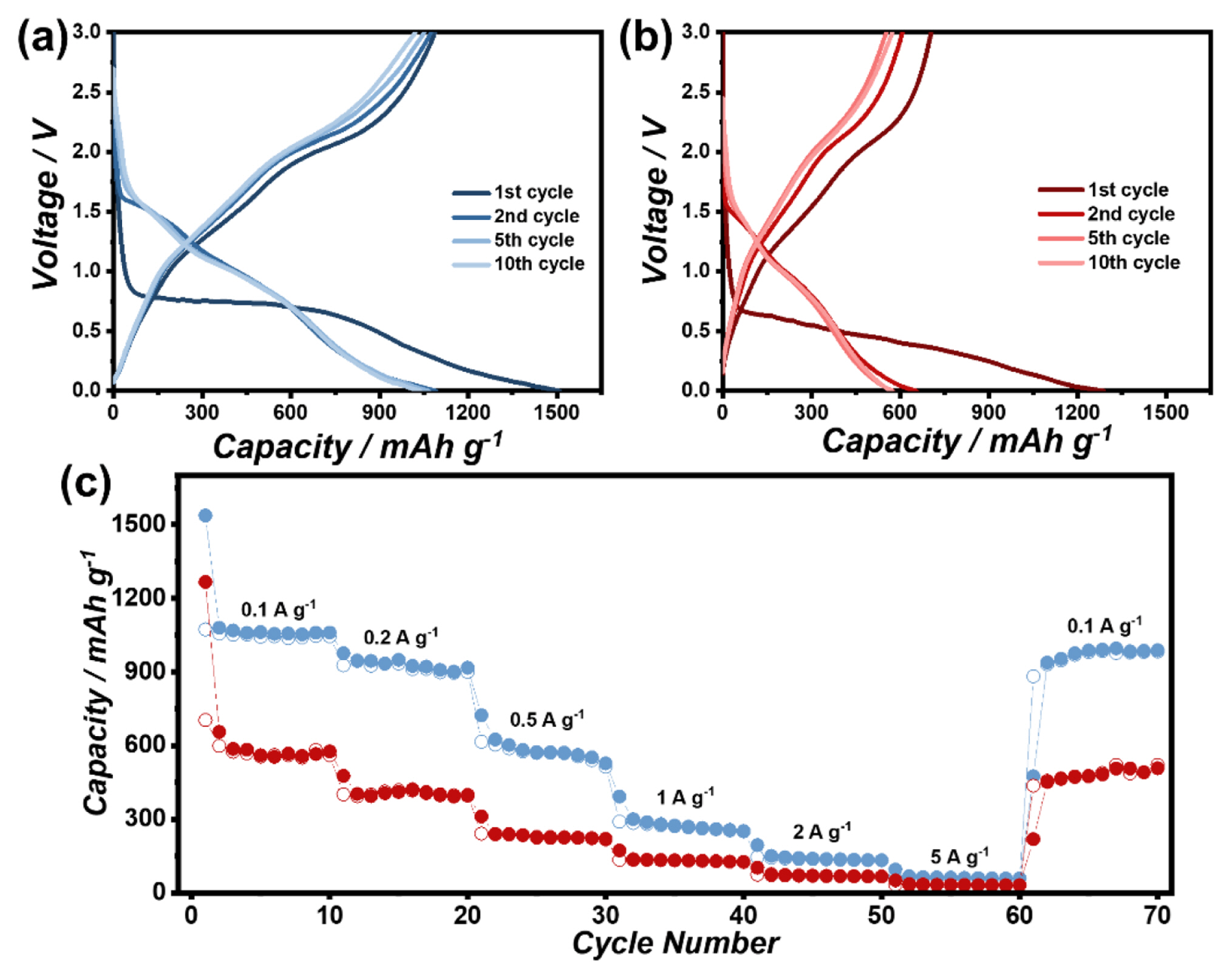
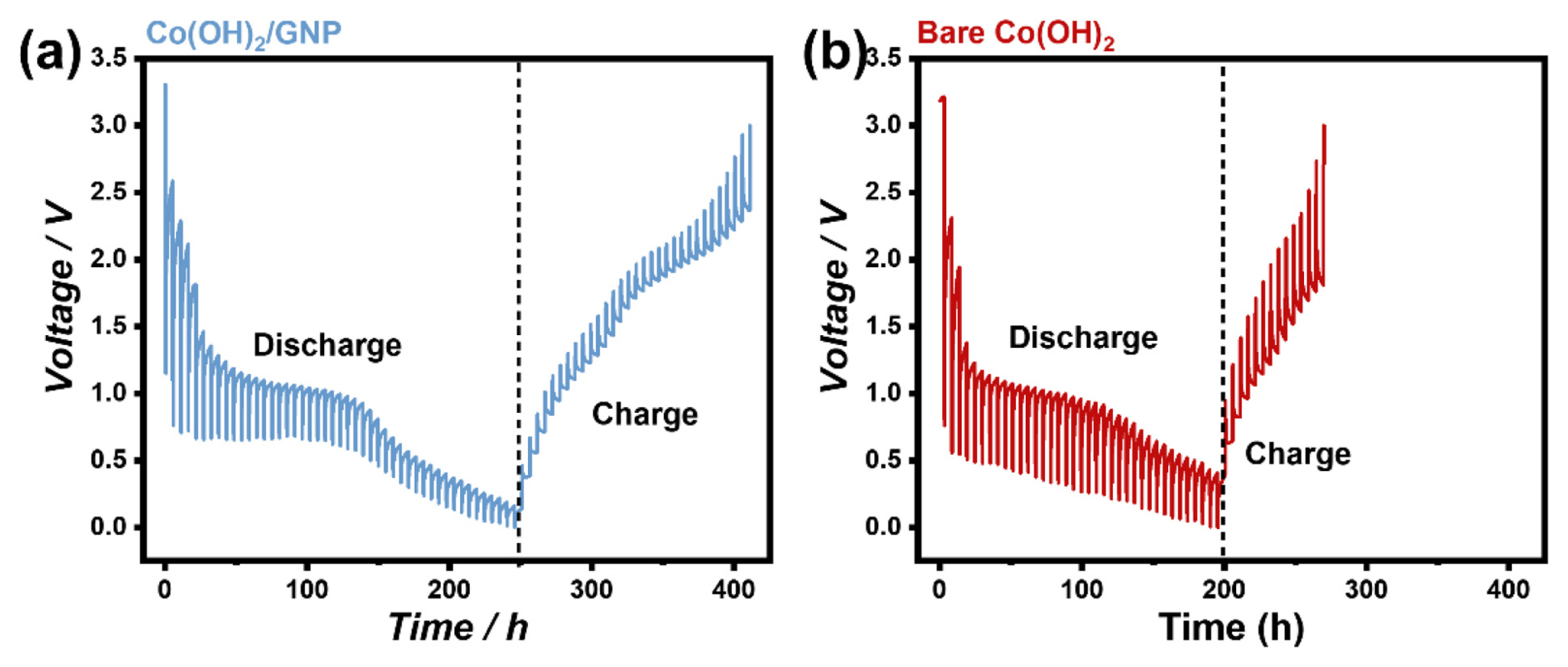
The electrochemical performance of the prepared materials was tested using galvanostatic cycling. Fig. 2(a) and (b) illustrate the voltage profiles of Co(OH)2/GNP and bare Co(OH)2 at a specific current of 100 mA g−1. According to the conventional conversion reaction mechanism, the theoretical capacity of Co(OH)2 is ~577 mAh g−1 (Co(OH)2 + 2 Li+ + 2 e− ↔ Co + 2 LiOH). The theoretical capacity of Co(OH)2/GNP can be calculated based on the TGA results and the maximum capacity of GNP as 744 mAh g−1 [13,24].
However, the observed capacities of the bare Co(OH)2 and Co(OH)2/GNP composites exceeded their theoretical values. The initial discharge and charge capacities were 1289 mAh g−1 and 705 mAh g−1 for bare Co(OH)2 and 1509 mAh g−1 and 1090 mAh g−1 for Co(OH)2/GNP, respectively. The large irreversible capacity loss during the first cycle could originate from the formation of a solid electrolyte interphase (SEI) layer [25,26], incomplete reoxidation of transition metals [27,28], and irreversible side reactions of inactive species [29]. The extra reversible capacity over the theoretical value during repeated cycling in the two materials can be ascribed to the redox reaction of LiOH [17,30]. Abnormal charge storage reactions, such as the formation/decomposition of the electrolyte-derived surface layer [31] and interfacial charge storage behaviors [32,33], could also contribute to the extra capacity, which are frequently observed in nanoscale conversion-based electrode materials [34–36]. Notably, the reversible capacity of 1090 mAh g−1 in Co(OH)2/GNP was ~1.8 times higher than the theoretical value and significantly superior to that of bare Co(OH)2. Furthermore, a rate capability test was conducted at different specific currents starting from 0.1 to 5 A g−1 with 10 cycles at each rate and returning to the initial rate, as depicted in Fig. 2(c). The specific capacities of both samples decreased as the current density increased. However, Co(OH)2/GNP composite still exhibited a high capacity of ~575 mAh g−1 at a specific current of 500 mA g−1, whereas only ~224 mAh g−1 was delivered by bare Co(OH)2. After returning to the initial rate of 100 mA g−1, Co(OH)2/GNP and bare Co(OH)2 showed reversible capacities of 942 mAh g−1 and 483 mAh g−1, at 86% and 74% of the second discharge capacities of each material, respectively, implying superior rate and cyclability of Co(OH)2/GNP composite materials.
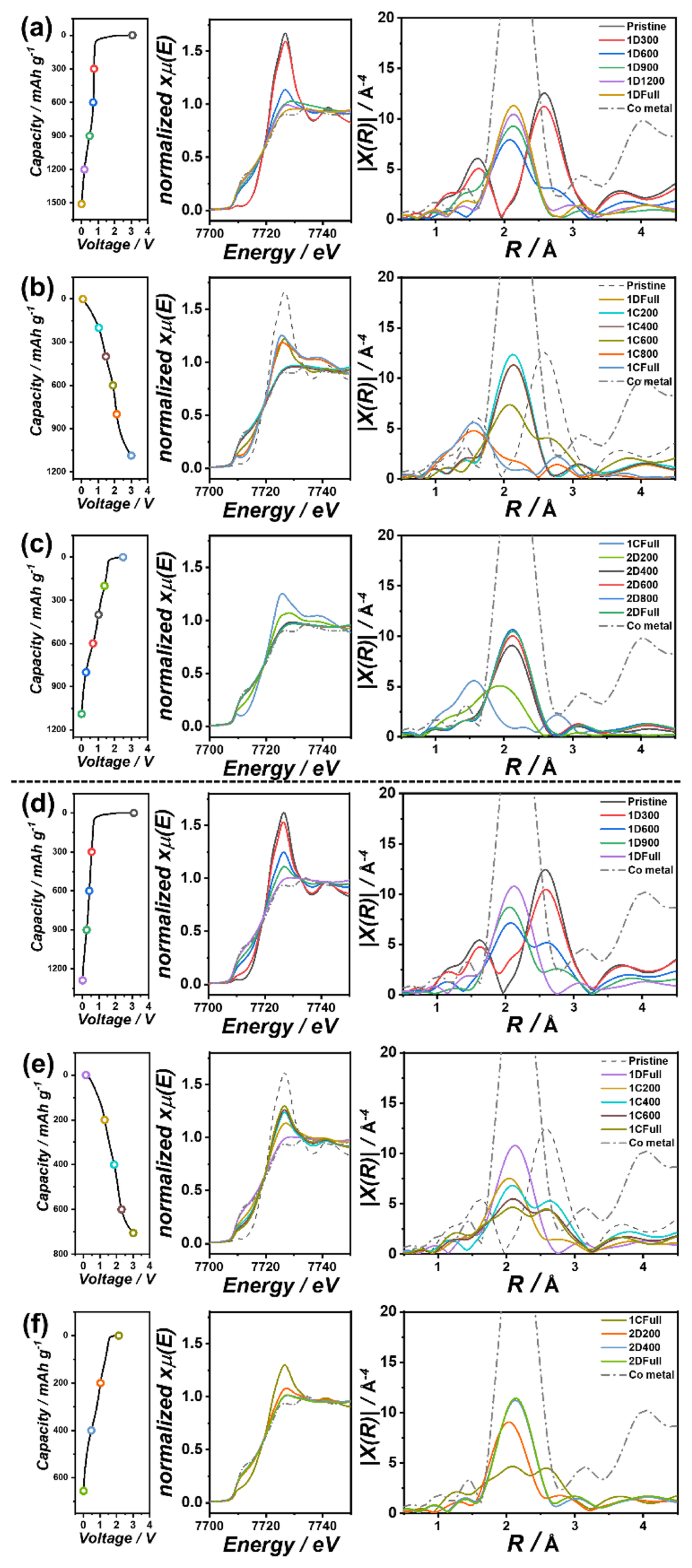
To investigate the detailed charge storage reaction of Co(OH)2/GNP, synchrotron-based XAS experiments were conducted, which is an efficient technique for observing nanoscale changes. Fig. 3(a) shows that the white line intensity and absorption edge position of the Co K-edge XANES spectra decreased during the first discharge process, indicating the reduction of Co2+ in the Co(OH)2 to Co0 state. In Fourier transformed EXAFS, the peaks corresponding to Co-O and Co-Co bonding of Co(OH)2 structure at 1.6 Å and 2.6 Å disappeared during the initial stage of the first discharge. Subsequently, a new peak at 2.1 Å appeared and increased, which corresponds to the Co metal phase. The XAS results during the first discharge process represent the breaking of the Co-O bond and the reduction of Co atoms to metallic Co due to the conversion reaction, which agrees with a previous report on nanostructured Co(OH)2 [17]. As shown in Fig. 3(b), the Co K-edge position shifted to a lower energy, and the Co metal peak in EXAFS decreased during subsequent charging. At the end of the charge, the Co-O bond peak at 1.6 Å increased again, indicating the reversibility of the conversion reaction. As depicted in Fig. 3(c), the changes occurred in reverse order at the second discharge. The overall variation trends in the XANES and EXAFS spectra of bare Co(OH)2 in Fig. 3(d–f) were similar to those of Co(OH)2/GNP.
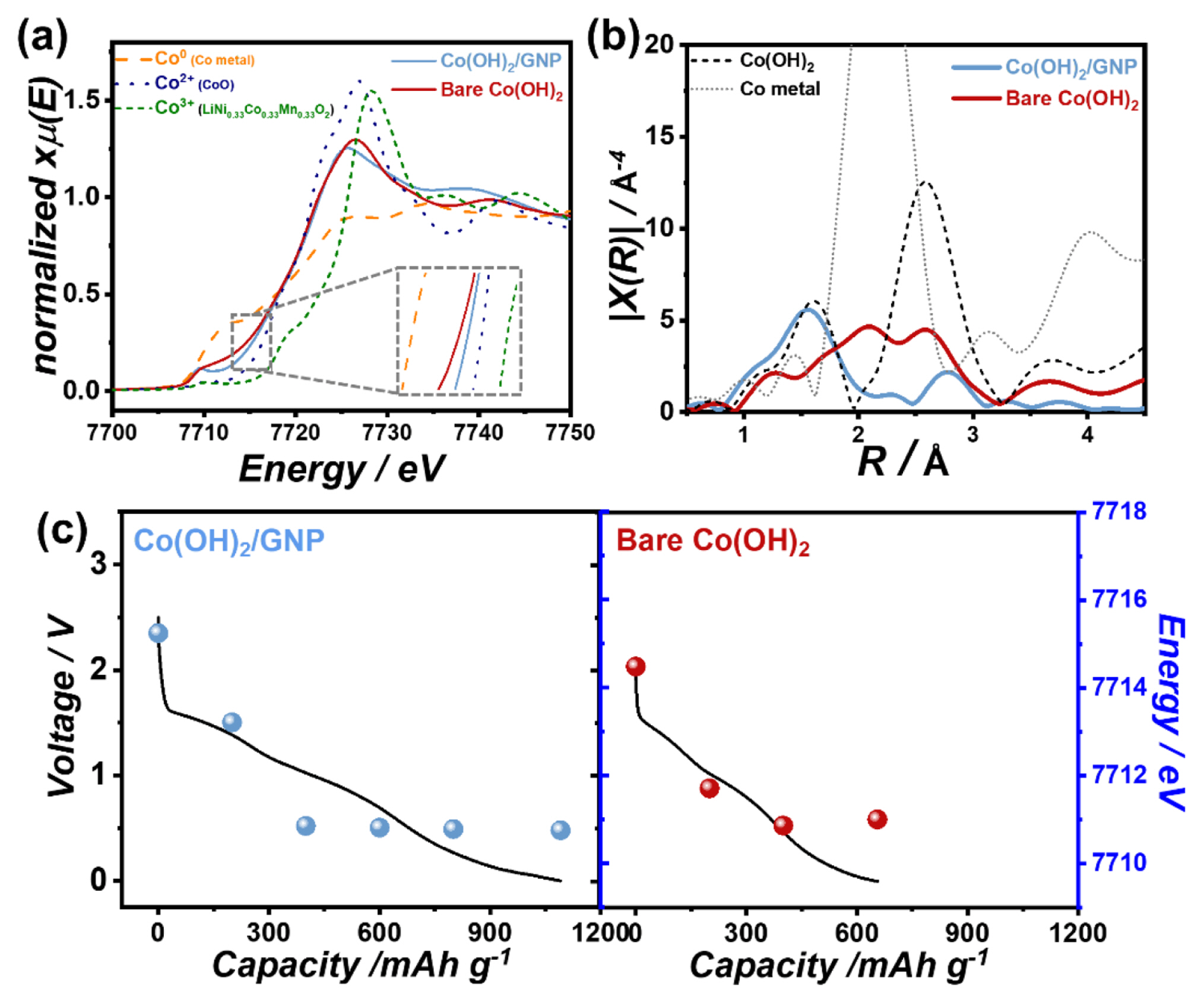
However, upon closer examination, the Co K-edge position after the fully charged state was higher for Co(OH)2/GNP than for Co(OH)2, as shown in Fig. 4(a), indicating that the recovery of the oxidation state of Co was more significant in the Co(OH)2/GNP electrode during delithiation. Additionally, the Co-O bond peak at 1.6 Å is noticeable in the Fourier transformed EXAFS of full charged Co(OH)2/GNP electrodes as illustrated in Fig. 4(b), while the Co metallic peak at 2.1 Å is still observed in bare Co(OH)2 after charge. The EXAFS spectrum of the fully charged bare Co(OH)2 electrode appears to be an intermediate state between the charged state of 600 mAh g−1 and the fully charged state of Co(OH)2/GNP. These results demonstrate that the reversibility of the conversion reaction was much greater for Co(OH)2/GNP. Furthermore, as shown in Fig. 4(c), the Co K-edge positions did not change noticeably during the discharge of Co(OH)2/GNP at a low potential range below ~1.0 V, although a large reversible capacity of over ~700 mAh g−1 was delivered in this potential range. In contrast, the capacity without changing the oxidation state of Co was only ~200 mAh g−1 in bare Co(OH)2. The minor changes in the Co K-edge position in the low-potential region indicate that the reversible capacity in the low-potential range did not originate from the conversion reaction. It has been reported that, in this potential region, the LiOH phase formed by the conversion reaction of transition metal hydroxides additionally reacts with lithium, delivering a large additional capacity [7,17,18]. Therefore, the large capacity in the low potential range during charging in Co(OH)2/GNP is ascribed to the high contribution of the reversible reaction of LiOH.
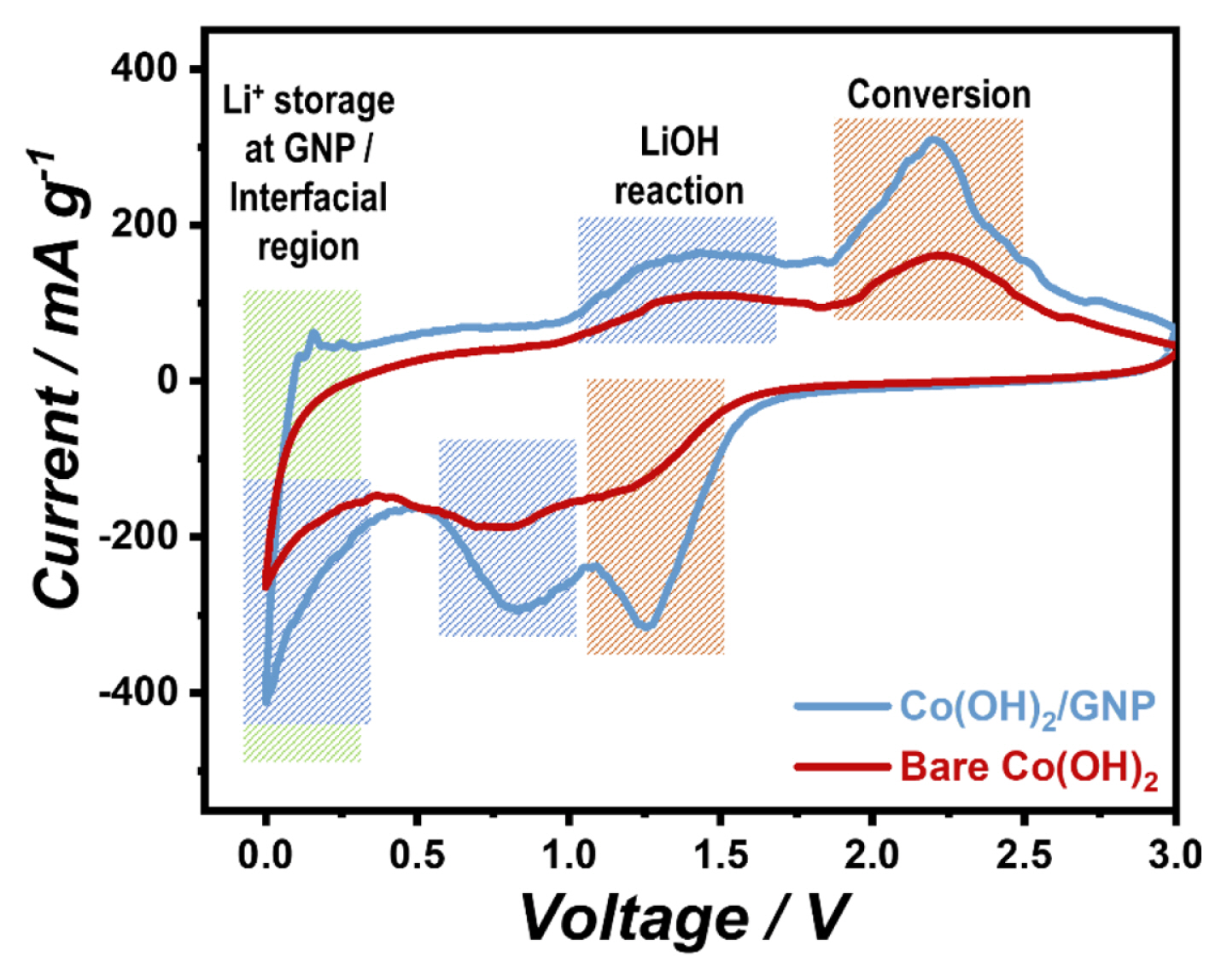
The above results demonstrate that the contribution of both the conversion and LiOH reactions is enhanced by the introduction of GNPs in Co(OH)2. To further verify the high contribution of these reactions, CV experiments were conducted, which is efficient for investigating electrochemical reactions occurring at a certain potential range [37]. As shown in the CV curves in Fig. 5, the peaks at ~2.20 V in anodic scan and ~1.25 V correspond to the conversion reaction. Other peaks from the reaction of the LiOH phase appeared at relatively low potentials, which are marked in the blue region [7,17,38]. Notably, the intensity of the peaks from the conversion and LiOH reactions in the CV curve of Co(OH)2/GNP is larger than that of bare Co(OH)2, which represents a greater contribution of the two reactions in the composite material. The facilitated conversion and LiOH reactions in the composite could be ascribed to the improved electronic conductivity and ionic diffusivity by introducing a conductive graphene matrix, resulting in faster reaction kinetics [13,39,40]. To confirm the beneficial effects of graphene, GITT experiments were conducted, as shown in Fig. 6. The GITT results demonstrate that the overpotential of Co(OH)2/GNP is significantly smaller than that of bare Co(OH)2 during electrochemical cycling, which corroborates the enhancement of the reaction kinetics in the composite [41]. At a potential close to 0 V (green region), the signals of the CV curves in Fig. 5 are also more prominent for Co(OH)2/GNP. The larger capacity of the composite in this potential range is ascribed to the ion storage behaviors of GNP [42] and facilitation of interfacial charge storage in the composite [43–45], as well as the increased LiOH reaction.
4. Conclusions
In this study, we comparatively analyzed the electrochemical performance of bare Co(OH)2 and Co(OH)2/GNP to investigate the beneficial effects of graphene on Co(OH)2-based composite materials. The prepared Co(OH)2/GNP exhibited a superior reversible capacity of 1090 mAh g−1, which is approximately 1.8 times higher than the theoretical value based on the conventional conversion reaction mechanism, whereas only ~705 mAh g−1 was delivered by bare Co(OH)2. Our experimental results demonstrate that the introduction of GNPs in the Co(OH)2-based anode material increases the electronic conductivity and diffusivity of the cation, thus facilitating the reversible conversion reaction and the reaction of the LiOH phase. Furthermore, additional lithium ions can be stored in the GNP and at the interfacial region. These results explain the improved electrochemical performance of the composite material. We anticipate that our results can provide new insights into lithium storage mechanisms beyond established reaction and design ideas for high-capacity electrode materials.