Electrochemical Oxidation of Amoxicillin in Its Commercial Formulation on Thermally Prepared RuO2/Ti
Article information
Abstract
In this work, a ruthenium dioxide electrode has been prepared by thermal decomposition at 400 ℃ then used for the oxidation of commercial amoxicillin. The physical characterization showed that RuO2 electrode presents a mud cracked structure. Its electrochemical characterization has revealed an increase of the voltammetric charge in acid electrolyte compared to neutral electrolyte indicating the importance of protons in its surface redox processes. The voltammetric study of the oxidation of amoxicillin has been investigated. It has been obtained that the oxidation of amoxicillin is controlled by both adsorption and diffusion processes. Moreover, the oxidation of amoxicillin occurs via direct and indirect processes in free or electrolyte containing chlorides. Through preparative electrolysis, enhancement of amoxicillin oxidation was observed in the presence of chloride where the amoxicillin degradation yield reached more than 50 % compared to less than 5% in the absence of chlorides. Spectrophotometric investigations have revealed the degradation of intermediates absorbing at 350 nm.
1. Introduction
In recent years, the situation of the pollution of our environment is alarming notably the water pollution [1,2]. That pollution is caused by agricultural activities, urban wastewater, industries wastewater and hospital wastewater which unfortunately are not treated. Among the large variety of pollutants that exist in the wastewaters, the hospital wastewater notably the pharmaceuticals are those which caused serious problems. The presence of pharmaceuticals in wastewater not only affects the water quality, but also causes long term potential adverse impacts on the ecosystems and the human health [3-5]. Among various pharmaceuticals, antibiotics are widely used in human and veterinary medicines to treat diseases and to promote growth [3,6]. The antibiotics can pose a potential environmental risk due to its extensive usage and resistance to natural biodegradation [7]. Since water is important for human life it becomes imperious to develop techniques that could be used to treat the antibiotics in hospital wastewaters. By now, it is important to indicate that no solution was found in our country to treat the hospital wastewater. To solve such a problem, biological degradation was very effective for the treatment of many organic compounds but it was not entirely efficient if non biodegradable compounds were present in the wastewaters. In that case, other techniques like Fenton reaction, ozonation, photocatalysis, sonication, irradiation or their combinations were used [8-13].
The choice of the type of the wastewater treatment to be used depends on the economics and also to the reliability and the treatment efficiency. Electrochemical methods have gained a great interest in that area because of the increased efficiencies that they can achieve using easy to operate and control electrochemical reactors. The electrochemical methods find several applications such as metal ion removal and recovery and destruction of toxic and non-biodegradable organics by direct or indirect anodic oxidation. In this technique, one of the problems was to find the best anodes that could be stable and present good electrocatalytic properties. Thus many studies have been carried out on the electrochemical treatment of organic compounds where several anode materials have been tested. In particular, the use of dimensionally stable anodes (DSA) for wastewater treatment has led to technological developments, thus reducing operational and investment costs. DSA electrodes exhibit high electrocatalytic activity, high stability to anodic corrosion, and excellent mechanical stability. But, by our knowledge, works dealing with the oxidation of the antibiotics especially the amoxicillin on DSAs were very scarce. Recently, Ti/IrO2-RuO2 and Ti/IrO2 have been used for the oxidation of pharmaceuticals such as tetracycline and oxacillin respectively [14,15]. Such investigations have shown that those organic compounds have been successfully degraded directly on the surface of those electrodes or via oxidative species such as hydroxyl radicals, chlorine which was generated during the oxidation processes [16,17]. Among DSAs, pure RuO2 was found to possess high electrochemical activity for the oxidation of various organics but has not been used for the degradation of amoxicillin. Thus in the present work, RuO2 was prepared by the thermal techniques and then characterized physically and electrochemically. The prepared electrode was used to carry out the electrochemical degradation of amoxicillin. The influence of chlorides on amoxicillin oxidation was also investigated.
2. Experiment
The electrode used in the following work was prepared in our laboratory with appropriate metallic precursor. The coating precursor was prepared from RuCl3, xH2O (Fluka). The precursor was dissolved in pure isopropanol (Fluka) used as solvent. The commercial products were used as received without any further treatment.
The titanium substrate on which the electrode films were deposited have the following dimension 1.6 cm × 1.6 cm × 0.5 cm for the voltammetric investigation and 5.0 cm × 5.0 cm × 0.5 cm for the preparative eletrolysis. The surface of each substrate was sandblasted to ensure good adhesion of the deposit on it. After sandblasting, the substrate was washed vigourously in water and then in isopropanol to clean their surface from residual sands. The substrate was then dried in an oven at 80 ℃ and weighed. After that, the precursor was applied by a painting procedure on cleaned titanium (Ti) substrate then put in an oven for 15 min at 80 ℃ to allow the evaporation of the solvent. Then after, it is put in a furnace at 400 ℃ for 15 min to allow the decomposition of the precursor. Theses steps were repeated until the desired weight of the coating is reached. A final decomposition of 1 h was done at 400 ℃. The deposit loading was about 5 g·m-2 on each Titanium substrates.
The physical characterization of the electrodes were performed using a scanning electronic microscopy (SEM, ZEISS, SUPRA 40VP) device.
The voltammetric measurements were performed on the prepared electrode in a three-electrode electrochemical cell using a Voltalab PJP 201. The counter electrode (CE) was a platinum wire and the reference electrode (RE) was a saturated calomel electrode (SCE). To overcome the potential ohmic drop, the reference electrode was mounted in a luggin capillary and placed close to the working electrode by a distance of 1 mm. The apparent exposed area of the working electrode was 1 cm2. For the exhaustive electrolysis, an ideally stirred batch reactor was used. The solution flow rate was 2.08 mL s-1. The mass transfer coefficient determined using the potassium ferri/ferrocyanide redox couple (Fluka) was 2.36×10-7 m s-1. The exposed electrode surface area with the solution was about 16 cm2. The other chemicals used in this work are composed of H2SO4 (Fluka), HClO4 (Fluka), KClO4 (Fluka), Na2SO4 (Fluka), NaCl (Fluka) and Amoxicillin tablets (from a pharmacy in Abidjan). All the chemicals have been used as received without any further treatment for the experiment. All the solutions used in the current work have been prepared with distilled water. All the experiments were made at ambient temperature of 25 ℃.
3. Results and Discussion
3.1 Physical characterization of the prepared electrode
In Fig. 1, the microphotos of the uncoated titanium plate (Fig. 1a, b) and the coated titanium plate (Fig. 1c) are presented. In Fig. 1a, the surface of titanium is smooth and presents a uniform and continuous structure. In Fig. 1b, the microphoto presents the sandblasted titanium plate. The surface of the sandblasted titanium looks different to that presented in Fig. 1a. The surface of the sandblasted titanium plate seems to be rough. In Fig. 1c, the prepared electrodes consisted in depositing RuO2 coating on the sandblasted titanium surface was presented. It appears from the comparison of the three microphotos that the titanium surface in Fig. 1c was totally covered by the deposit. The surface of the electrode is rough with mud cracked structure. Pores are also present on the deposit.

Scanning electron micrograph of: (a) smooth titanium, (b) sandblasted titanium and (c) RuO2 electrode.
3.2 Electrochemical characterization of the electrode in the potassium perchlorate medium
The cyclic voltammetry measurements realized in potassium perchlorate at several potential scans rates on the electrode are presented in the Fig. 2a. In this figure, the results of these investigations showed that the voltammograms have all the same feature. In case of the measurement realized under a scan rate of 6 mVs-1, the voltammogram showed that the current density is almost constant from -0.5 to 0.4 V in the forward potential scan. From 0.4 to 0.9 V an increase of the current density is observed. An oxidation wave is observed around 0.78 V. The oxygen evolvement was observed by the rapid increase in the current density after 0.9 V. On the backward potential scan, an oxide reduction wave is observed around -0.2 V. From those results, it appears that the reversible redox transition peaks usually observed in the acidic electrolytes in the potential domain starting from 0.15 V vs. SCE to 0.4 V vs. SCE are absent.
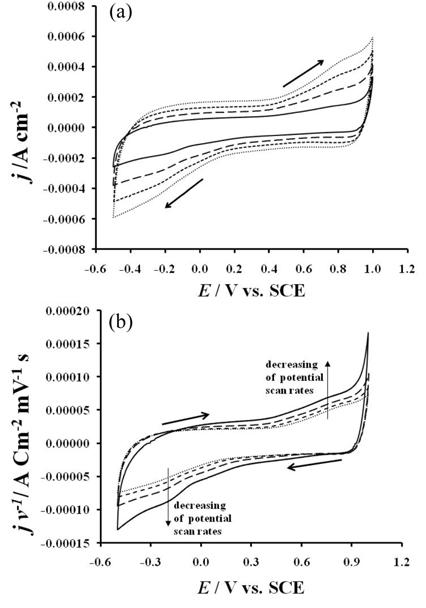
Cyclic voltammetry investigation in KClO4 0.1 M on the electrode (a): at different scans rates: 2 mV s-1  , 4mV s-1
, 4mV s-1  , 6mV s-1
, 6mV s-1  , 8mV s-1
, 8mV s-1  , (b): normalized current density with the potential scan rates versus applied potential (jv-1-E); CE: platinum wire, RE: SCE, T = 25 ℃.
, (b): normalized current density with the potential scan rates versus applied potential (jv-1-E); CE: platinum wire, RE: SCE, T = 25 ℃.
Therefore, the presence of the anodic wave at 0.78 V followed by its counterpart at -0.2 V in the cathodic region of the voltammogram is characteristic of an irreversible surface transition process. This finding reveals that protons play an important role in the reversibility of the electrode surface redox transition processes. As it is also seen in Fig. 2a, the voltammetric charge decreases when the potential scan rates decrease. When the current densities are normalized with the potential scan rates and plotted against the applied potential, Fig. 2b is obtained. In this figure, the curves present a symmetrical aspect. The discharged current recorded in the negative going potential scan is entirely recovered in the positive going potential scan. In this figure, as the potential scan rates increases, the capacitance decreases indicating that for the low potential scan rates, more electrode active sites intervene in the surface processes. Indeed, at the low potential scan rates, the electrolyte penetrates into the pores and reaches the prepared electrode’s inside active sites. The slightly increase of the capacitance at E > 0.6 V and at E < -0.1 V could be related to the sluggish processes occurring at the electrode surface.
In Fig. 3, the voltammograms recorded in various electrolytes are presented. This figure showed that the voltammetric charge is higher in acid medium (HClO4, H2SO4) than in neutral medium (KClO4, Na2SO4). Thus, the presence of the protons H+ allows increasing the voltammetric charge and also the conductivity of the electrolyte. The increase in the voltammetric charge could result from the surface redox processes. More active sites react with the protons in such processes. The cyclic voltammetry measurement realized on RuO2 electrode in the sulfuric or perchloric acid electrolyte presents the general characteristics of RuO2 electrode. This voltammogram has a rectangular shape with waves of low current density at 0.35 V and 0.88 V. According to literature [18-20], the presence of these waves could be due respectively to the redox transitions of Ru (III) / Ru (IV) and Ru (IV) / Ru (VI) scanning at the surface of the electrode. A rapid increase of the current related to OER started at 1.05 V. The current density of the oxygen evolvement in the acid medium is almost vertical. This result showed that the protons H+ contribute significantly in the reduction of the ohmic drop of the electrolyte by increasing the conductivity of the supporting electrolyte.
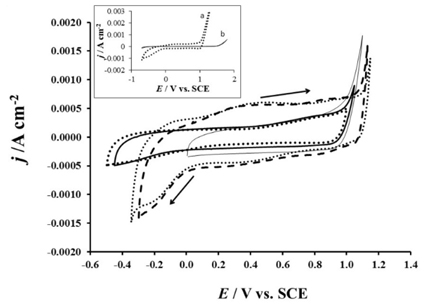
Cyclic voltammograms on RuO2 electrode in KClO4 0.1 M  , Na2SO4 0.1 M
, Na2SO4 0.1 M  , HClO4 0.1 M
, HClO4 0.1 M  H2SO4 0.1 M
H2SO4 0.1 M  , and in KClO4 0.1 M + NaCl 100 mg L-1
, and in KClO4 0.1 M + NaCl 100 mg L-1  , in the inset: voltammetric investigation of (a) RuO2 and (b) Ti electrodes in KClO4 0.1 M. CE: platinum wire, RE: SCE, 6 mV s-1 , T = 25 ℃
, in the inset: voltammetric investigation of (a) RuO2 and (b) Ti electrodes in KClO4 0.1 M. CE: platinum wire, RE: SCE, 6 mV s-1 , T = 25 ℃
In Fig. 3, the results of the cyclic voltammetry measurement realized on the electrode in KClO4 electrolyte and in KClO4 electrolyte containing chlorides are also presented. This figure showed that the voltammograms have practically the same feature in the two electrolytes. However in KClO4 electrolyte containing chlorides, the onset of the potential related to the increase of the current density (0.89 V) is lower than that of the oxygen evolution reaction (OER) potential (1.05 V). The increase of the current density at 0.89 V in the chloride containing electrolyte could be due to the chlorine evolvement. In the inset of Fig. 3, cyclic voltammetry measurements performed on sandblasted titanium and RuO2 coated titanium were presented. The obtained results revealed that the voltammetric charge related to the electrode active sites is almost zero on Ti conversely to the high voltammetric charge recorded with RuO2. Moreover, the oxygen evolution reaction seems to be very sluggish on Ti than on RuO2 since the onset of the potential of OER are respectively 1.55 V and 1.05 V.
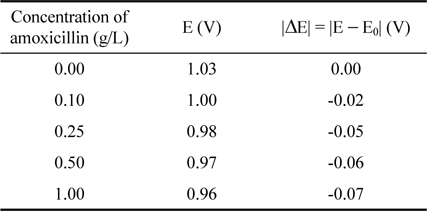
Fig. 4a presents the voltammograms realized on the electrode in the absence and presence of amoxicillin. The current density starts to increase after 0.47 V in the presence of amoxicillin. This result indicates that the oxidation of amoxicillin starts at 0.47 V in the domain of water stability. In the high anodic potential domain, a decrease of the overpotential of oxygen evolution is observed in the presence of amoxicillin. For a given current density of 0.005 A cm-2, the amoxicillin oxidation potentials have been recorded for several concentrations and assigned in the Table 1. The gap |ΔE| between potentials registered in the presence and absence of amoxicillin have been calculated and also reported in the table 1. The shift of the oxygen evolution overpotential in the negative direction became important as the concentration of the amoxicillin increases. That observation indicated that the intermediates resulted from water decomposition are involved in the amoxicillin oxidation in that domain. Moreover, this process occurs in the domain where the surface redox transition process takes place indicating that the oxidation of the amoxicillin occurs on oxidized ruthenium dioxide surface.
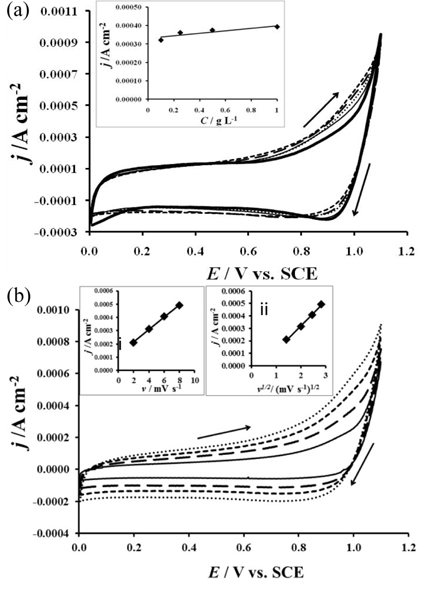
Cyclic voltammetry investigation (a): of amoxicillin in KClO4 for different concentrations of amoxicillin (0 g L-1  ; 0.1 g L-1 ; 0.25 g L-1 ; 0.5 g L-1 ; 1 g L-1 ) on the electrode at 8 mV s-1 and in the inset: (i) Evolution of the anodic waves current density against amoxicillin concentration at E= 0, 90 V. (b): of 1 g L-1 amoxicillin at different scans rates: 2 mV s-1 , 4 mV s-1 , 6 mV s-1 , 8 mV s-1 and in the inset: (ii) Evolution of the oxidation wave current density at 0.95 V against the scan rate and against square root of potential scan rates. CE: platinum wire, RE: SCE, T=25 ℃
; 0.1 g L-1 ; 0.25 g L-1 ; 0.5 g L-1 ; 1 g L-1 ) on the electrode at 8 mV s-1 and in the inset: (i) Evolution of the anodic waves current density against amoxicillin concentration at E= 0, 90 V. (b): of 1 g L-1 amoxicillin at different scans rates: 2 mV s-1 , 4 mV s-1 , 6 mV s-1 , 8 mV s-1 and in the inset: (ii) Evolution of the oxidation wave current density at 0.95 V against the scan rate and against square root of potential scan rates. CE: platinum wire, RE: SCE, T=25 ℃
ΔE against amoxicillin concentration (E0 = 1.03 V)
Additionally the current density recorded at 0.95 V increases as the concentration of the amoxicillin increases. Those current densities plotted against the concentrations of amoxicillin showed a linear evolution (inset of Fig. 4a) indicating that the appearance of those current densities was directly related to the amoxicillin oxidation.
Fig. 4b showed that the voltammetric charge increases with the potential scans rates. The current density recorded at 0.95 V and plotted against the potential scan rates is presented in the inset of Fig. 4b. A linear evolution is observed indicating that the oxidation of amoxicillin process occurs via adsorption (Fig. 4b-i). The plot of the current density recorded at 0.95 V against the square root of the potential scan rates led to a straight line indicating that the process is diffusion controlled (Fig. 4b-ii). A mixed control process such as adsorption and diffusion occurs during the oxidation of amoxicillin on the RuO2 electrodes.
Fig. 5 presented the results of the cyclic voltammetry measurement realized on the electrode in KClO4 containing amoxicillin in the absence or in the presence of chlorides. In this figure, one observed that in the presence of chlorides, the voltammetric charge increases and the current density starts being rapid at 0.98 V. The obtained results clearly show that the chlorides contribute in enhancing the kinetic of the amoxicillin oxidation. In fact, the higher current density recorded in the presence of chlorides could result from the oxidation between Cl2 and the amoxicillin leading to the generation of oxidized by product on the electrode surface.
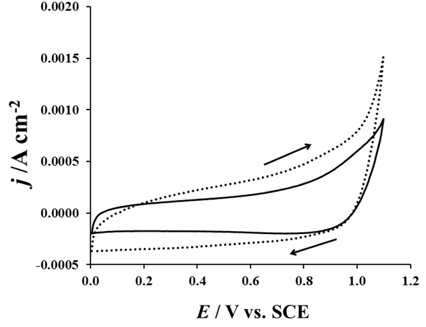
Cyclic voltammetry of the electrode in KClO4 0.1 M + amoxicillin 1g L-1 and in KClO4 0.1 M + amoxicillin 1 g L-1 containing NaCl 0.1 g L-1 ; v = 6 mV s-1, CE: platinum wire, RE: SCE, T=25 ℃.
3.3 Electrolysis of the amoxicillin
The electrolysis of amoxicillin has been performed in a batch reactor that was ideally stirred. In this experiment, a neutral and an acidic electrolyte have been investigated. Fig. 6 presents the curves resulting from the electrolysis of amoxicillin under 20 mA cm-2. In both the solutions, the chemical oxygen demand (COD), a global parameter, has been followed. To allow an easier comparison between the obtained results, normalized COD* (1) a dimensionless parameter, has been calculated and presented in Fig. 6. One observed that the COD* decreases with time.
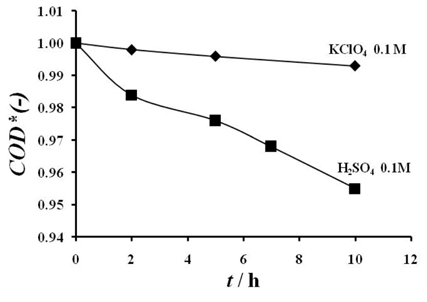
Electrolysis of 1 g L-1 of amoxicillin on RuO2 electrode in KClO4 0.1 M and H2SO4 0.1 M at 20 mA cm-2. Anode: RuO2, cathode: Titane, T = 25 ℃.

Where COD0 and CODt corresponds to the chemical oxygen demand at t =0 and t ≠ 0.
The decrease of the COD* is very low in both the electrolytes since the degradation yield calculated after 10 h of electrolysis is about 0.4 % and 4.53 % in potassium perchlorate and in sulfuric acid respectively. Meanwhile, it seems to be more rapid in sulfuric acid than in potassium perchlorate. In chlorides containing electrolytes, the electrolysis of amoxicillin has been investigated and the obtained results were presented in Fig. 7. Those results showed that in the presence of chlorides, the oxidation kinetic of amoxicillin increases as the chlorides concentration increases.
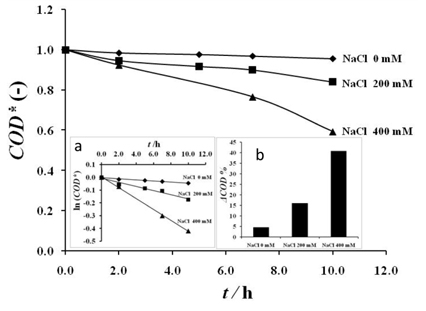
Electrolysis of 1 g L-1 Amoxicillin on RuO2 electrodes in H2SO4 0.1 M containing several concentration of NaCl at 20 mA cm-2; Anode: RuO2; cathode: Titane, T = 25 ℃; in the inset: (a) plot of ln(COD) versus time and (b) plot of ΔCOD % versus NaCl concentration
In the inset of that figure, the evolution of the logarithm of the chemical oxygen demand versus the electrolysis time was presented. Straight lines were obtained indicating that the oxidation process followed a pseudo first order kinetic (Fig. 7a). In the inset of Fig. 7, the removal yield of the chemical oxygen demand versus the concentration of the sodium chloride is also presented (Fig. 7b). These results revealed that the COD removal yields are 4.53, 15.99 and 40.71 respectively in the electrolytes containing 0 mM, 200 mM and 400 mM of NaCl after 10 h of electrolysis. According to the obtained results, one can indicate that amoxicillin can be degraded via a catalytic oxidation mechanism that involved a direct oxidation of amoxicillin on the electrode surface and/or by an indirect oxidation via chlorine species and possibly via other in situ redox species. In fact the direct electrooxidation and the electrooxidation mediated by hydroxyl radicals or by a higher oxide occurred according to the cyclic voltammetry investigation in the absence of chlorides. Under the applied current density, water undergoes discharged over the RuO2 electrode surface leading to the formation of hydroxyl radicals that immediately contribute to the oxidation of the RuO2 surface forming a higher oxide RuO3. In the presence of chlorides, the following mechanism could also take place during the electrolysis process [8]. In a first step, chloride ions are oxidized into chlorine at the RuO2 anode surface according to the reaction

The second step was the formation of hypochlorous acid
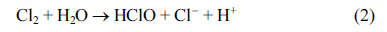
The HClO undergoes dissociation into hypochlorite and hydrogen ions:

Then indirect oxidation of the amoxicillin by the hypochlorite occurred either in the vicinity of the electrode surface or in the bulk of the solution.
During the electrolysis, the absorbance of the samples withdrawn from the simulated wastewater tank containing a solution of amoxicillin without chlorides was recorded. The obtained result is shown in Fig. 8a-b. In the inset of Fig. 8a, the peak of the absorbance recorded at about 350 nm was plotted against the electrolysis time (Fig. 8a-i). In Fig. 8a-i, one observes that during the first 5 h, an increase of the absorbance is observed. After 5 h, the absorbance was practically constant. That could be due to the production of intermediates which absorbed at the same wavelength as amoxicillin probably intermediates which molecular structures are similar to the parent amoxicillin. In this experimental condition and after 5h, the removal of the organic species present in the solution seems difficult to occur probably because of the fact that the surface of the electrode was totally covered by adsorbed species that decreases consequently the production of hydroxyl radical which would paticipate in the degradation of amoxicillin and its intermediates.
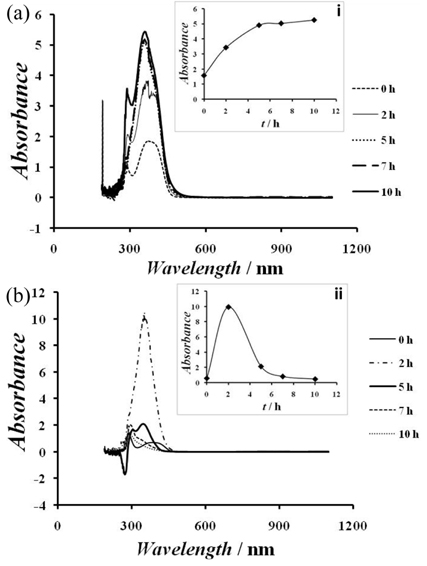
Evolution of the absorbance of amoxicillin versus wavelength during the electrolysis of amoxicillin at 20 mA cm-2 on RuO2 electrodes (a): in the absence of NaCl and in the inset: (i) Evolution of the absorbance against electrolysis time (wavelength = 350 nm). (b): (ii) in the presence of 400 mM NaCl and in the inset: Evolution of the absorbance against electrolysis time (wavelength = 350 nm).
The absorbance of the samples withdrawn from the simulated wastewater tank was also recorded in the amoxicillin electrolyte containing NaCl 400 mM. The obtained result is shown in Fig. 8b. In Fig. 8b-ii, the peak of the absorbance was plotted against the electrolysis time. This figure showed that during the first 2 h of electrolysis, the absorbance increases and reaches a maximum value. That was then followed by a decrease of the absorbance tending towards zero absorbance after 10 h of electrolysis. That observation indicates that during the electrolysis of amoxicillin, intermediates are produced and their concentration increases until the first 2 h. After 2h, the removal of intermediates absorbing around 350 nm occurs. After 10 h of electrolysis, a total degradation of the amoxicillin and those intermediates was reached. In fact in the presence of chloride, chlorine formation seems to be more rapid than the organic adsorption process so that they undergo oxidation by chlorine in the vicinity of the electrode or in the bulk of the solution as soon as they are produced. The production of chlorine enhances the RuO2 electrode degradation performance.
4. Conclusion
The prepared RuO2 electrode presents a mud cracked structure. The voltammetric investigation in acid and neutral electrolytes showed that protons play an important role in enhancing the electrolyte conductivity and also favour the electrode surface redox transition processes. Thus in acid electrolyte, the voltammetric charge of the electrode increases comparatively to neutral electrolyte. Amoxicillin undergoes oxidation on the Ruthenium dioxide. That oxidation is controlled by adsorption and diffusion processes in perchlorate electrolyte free of chloride. In the presence of chlorides, an enhancement of that compound oxidation occurs through the increase in the current intensity and the decrease of the onset of the gas evolvement potential. The oxidation of amoxicillin occurs also via a mixed oxidation processes involving direct and indirect routes either in KClO4 electrolyte free of chlorides or in chlorides containing perchlorate electrolyte. The exhaustive or preparative electrolysis of amoxicillin has revealed that enhancement of its oxidation is improved in presence of chlorides and that favour a total oxidation of intermediates having the same molecular structure as amoxicillin.
Acknowledgements
We greatly thank the Swiss National Funds for its financial support that allowed this work to be carried out. Our Team has received part of the grant IZ01Z0_146919 for that work. We also thank Prof. Bakayoko-Ly Ramata, the President of the University Felix Houphouet-Boigny for her help in the realization of that work.
References
K. M. Yao, B. S. Métongo, A. Trokourey and Y. Bokra, J. Océanol. Limnol. Abidjan, IV, 1 (2007).
Yao K. M., Métongo B. S., Trokourey A., Bokra Y.. J. Océanol. Limnol. Abidjan (2007);IV:1.P. L. A. Guillaume, K. H. Honoré, T. Albert and L. Ouattara, Int. J. Pure Appl. Sci. Technol., 14(1), 33 (2013).
Guillaume P. L. A., Honoré K. H., Albert T., Ouattara L.. Int. J. Pure Appl. Sci. Technol. (2013);14(1):33.N. Oturan, J. Wu, H. Zhang, V. K. Sharma and M. A. Oturan, Appl. Catal. B: Environmental, 140-141, 92 (2013).
Oturan N., Wu J., Zhang H., Sharma V. K., Oturan M. A.. Appl. Catal. B: Environmental (2013);140-141:92. 10.1016/j.apcatb.2013.03.035.I. Sirés and E. Brillas, Environment International, 40, 212 (2012).
Sirés I., Brillas E.. Environment International (2012);40:212. 10.1016/j.envint.2011.07.012.J. R. Domínguez, T. González, P. Palo and J. Sánchez-Martín, Chem. Eng. J., 162, 1012 (2010).
Domínguez J. R., González T., Palo P., Sánchez-Martín J.. Chem. Eng. J. (2010);162:1012. 10.1016/j.cej.2010.07.010.P. Wang, P.-S. Yap and T.-T. Lim, Appl. Catal. A. Gen., 399, 252 (2011).
Wang P., Yap P.-S., Lim T.-T.. Appl. Catal. A. Gen. (2011);399:252. 10.1016/j.apcata.2011.04.008.H. Lin, J. Niu, J. Xu, Y. Li and Y. Pan, Electrochim. Acta, 97, 167 (2013).
Lin H., Niu J., Xu J., Li Y., Pan Y.. Electrochim. Acta (2013);97:167. 10.1016/j.electacta.2013.03.019.I. Tantis, M. Antonopoulou, I. Konstantinou and P. Lianos, J. Photochem. Photobiol. A Chem., 317, 100 (2016).
Tantis I., Antonopoulou M., Konstantinou I., Lianos P.. J. Photochem. Photobiol. A Chem. (2016);317:100. 10.1016/j.jphotochem.2015.11.013.M. Solak, M. Kílíç, Y. Hüseyin and A. Sencan, J. Hazard. Mater., 172, 345 (2009).
Solak M., Kılıç M., Hüseyin Y., Sencan A.. J. Hazard. Mater. (2009);172:345. 10.1016/j.jhazmat.2009.07.018.I. R. Bautitz and R. F. P. Noguiera, J. Photochem. Photobiol. A Chem., 187, 33 (2007).
Bautitz I. R., Noguiera R. F. P.. J. Photochem. Photobiol. A Chem. (2007);187:33. 10.1016/j.jphotochem.2006.09.009.J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 36, 1 (2006).
Pignatello J. J., Oliveros E., MacKay A.. Crit. Rev. Environ. Sci. Technol. (2006);36:1. 10.1080/10643380500326564.W. Hua, E. R. Bennett and R. J. Letcher, Water Research, 40, 2259 (2006).
Hua W., Bennett E. R., Letcher R. J.. Water Research (2006);40:2259. 10.1016/j.watres.2006.04.033.R. Hernandez, M. Zappi, J. Colucci and R. Jones, J. Hazard. Mater., 92, 33 (2002).
Hernandez R., Zappi M., Colucci J., Jones R.. J. Hazard. Mater. (2002);92:33. 10.1016/S0304-3894(01)00371-5.E. A. Serna-Galvis, J. Silva-Agrego, A. L. Giraldo, O. A. Florez and R. A. Torres-Palma, Chem. Eng. J., 284, 953 (2016).
Serna-Galvis E. A., Silva-Agrego J., Giraldo A. L., Florez O. A., Torres-Palma R. A.. Chem. Eng. J. (2016);284:953. 10.1016/j.cej.2015.08.154.J. Wu, H. Zhang, N. Oturan, Y. Wang, L. Chen and M. A. Oturan, Chemosphere, 87, 614 (2012).
Wu J., Zhang H., Oturan N., Wang Y., Chen L., Oturan M. A.. Chemosphere (2012);87:614. 10.1016/j.chemosphere.2012.01.036.A. Rubio-Clemente, R. A. Torres-Palma and G. A. Penuela, Sci. Total Environ., 478, 201 (2014).
Rubio-Clemente A., Torres-Palma R. A., Penuela G. A.. Sci. Total Environ. (2014);478:201. 10.1016/j.scitotenv.2013.12.126.F. L. Souza, J. M. Aquino, D. W. Miwa, M. A. Rodrigo and A. J. Motheo, J. Environ. Chem. Eng., 2, 811 (2014).
Souza F. L., Aquino J. M., Miwa D. W., Rodrigo M. A., Motheo A. J.. J. Environ. Chem. Eng. (2014);2:811. 10.1016/j.jece.2014.02.003.R. Manoharan and J. B. Goodenough, Electrochim. Acta, 36, 19 (1991).
Manoharan R., Goodenough J. B.. Electrochim. Acta (1991);36:19. 10.1016/0013-4686(91)85174-6.A. V. Rosario, L. O. S. Bulhões and E. C. Pereira, J. Power Sources, 158, 795 (2006).
Rosario A. V., Bulhões L. O. S., Pereira E. C.. J. Power Sources (2006);158:795. 10.1016/j.jpowsour.2005.09.002.A. Kapalka, G. Fóti and C. Comninellis, Electrochem. Commun., 10, 607 (2008).
Kapalka A., Fóti G., Comninellis C.. Electrochem. Commun. (2008);10:607. 10.1016/j.elecom.2008.02.003.