Electrochemical Characterization of Electric Double Layer Capacitors Assembled with Pyrrolidinium-Based Ionic Liquid Electrolytes
Article information
Abstract
We present the electrochemical performance of electric double layer capacitors (EDLCs) assembled with pyrrolidinium (Pyr)-based ionic liquid electrolytes at 55 ℃. Cations with various alkyl chain lengths were employed in Pyr-based ionic liquids to investigate the effect of cation structure on the cycling stability of EDLCs. The EDLCs exhibited initial specific capacitances ranging from 122.4 to 131.6 F g−1 based on activated carbon material at 55 ℃. Cycling data and XPS results demonstrate that Pyr-based ionic liquid with longer alkyl chain is more effective for enhancing the cycling stability of EDLC by suppressing the reductive decomposition of pyrrolidinium cations during cycling at high temperatures.
1. Introduction
Ionic liquids are molten salts with their melting points close to room temperature. They are generally composed of a bulky, asymmetric, and organic cation and a weakly-coordinating inorganic anion [1-3]. Ionic liquids have attractive properties such as negligible vapor pressure, wide electrochemical stability, high ionic conductivity, non-flammability and high thermal stability [4-6]. These properties make them promising candidates for use in electrolytes for electrochemical devices, including rechargeable lithiumion batteries and electric double layer capacitors (EDLCs) with enhanced safety [7-9]. EDLCs have been considered one of the most promising electrochemical energy storage devices with high specific power and long cycle life [10-12]. They can be used in power electronics for back-up memory, hybrid electric vehicles and other energy storage systems that cannot be connected to conventional power suppliers, such as windmills. In hybrid electric vehicles, EDLCs can be coupled with fuel cells or rechargeable batteries to deliver the high power needed during acceleration as well as to recover energy during braking [13]. ELDCs operating at high temperatures are needed for various applications such as the replacement of electrolytic capacitors. In ELDCs, the charge storage is electrostatic and the electrolyte ions are reversibly adsorbed in the electric double layer of the porous carbon electrode [14]. Most EDLCs on the market use electrolyte solutions based on aprotic solvents, such as acetonitrile and carbonated propylene, and operate at room temperature [15]. However, the use of organic electrolytes poses safety issues arising from the high flammability of organic solvents [16]. Furthermore, EDLCs employing organic electrolytes cannot be operated at high temperatures because the electrochemical stability of organic solvents decreases with increasing temperature, resulting in significant reduction of cycle life [17,18]. In order to overcome these problems, the use of ionic liquids as electrolytes in EDLCs for high temperature applications has been investigated [19-23]. Among various ionic liquid electrolytes, 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIBF4) has received considerable attention as a promising electrolyte in EDLC due to its low viscosity and high ionic conductivity [24,25]. However, imidazolium-based ionic liquids typically exhibit poor reductive stability at lower potentials, and thus it is reductively decomposed on the negative electrode during cycling of EDLCs with high operating voltages [26]. Pyrrolidinium (Pyr)-based ionic liquids can be an alternative ionic liquid electrolyte for EDLC applications due to their high electrochemical stability and hydrophobic nature [27,28].
In this work, we investigate and discuss the electrochemical performance of EDLCs assembled with Pyr-based ionic liquid electrolytes at 55 ℃. In Pyr-based ionic liquids, cations with various chemical structures were employed to investigate the effects of cation structure on the cycling stability of EDLC because the cation affects the reductive stability. Our results demonstrate that Pyr-based ionic liquid with longer alkyl chains is more effective in enhancing the cycling stability of EDLC by suppressing the reductive decomposition of pyrrolidinium cations during cycling at high temperature.
2. Experimental Section
2.1. Cell assembly
We used three different ionic liquids, 1-methyl-1-propylpyrrolidinium bis(trifluoromethanesulfonyl) imide (Pyr13-TFSI), 1-methyl-1-butylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr14-TFSI), and 1-methyl-1-pentylpyrrolidinium bis(trifluoromethanesulfonyl) imide (Pyr15-TFSI); their molecular structures are shown in Fig. 1. The ionic liquids were purchased from Solvionic and used after drying under vacuum at 120 ℃ for 24 h. The water content in the ionic liquids after vacuum drying was less than 20 ppm via Karl Fisher titration. The electrode was prepared by coating an N-methyl pyrrolidone (NMP)-based slurry containing activated carbon powder (Power Carbon Technology), polytetrafluoroethylene (PTFE), poly (vinyl pyrrolidone) (PVP), and super-P carbon (MMM Co.) (80:5:5:10 by weight) onto aluminum foil. The resulting electrodes were dried in a vacuum oven at 110 ℃ for 12 h. The thickness of the electrode was approximately 70 μm. The EDLC was assembled by sandwiching a nonwoven cellulose separator (TF4035, NKK) between two symmetric carbon electrodes. The cell was then enclosed in a CR2032-type coin cell and injected with various ionic liquid electrolytes (Pyr13-TFSI, Pyr14-TFSI, Pyr15-TFSI). All cells were assembled in a dry box filled with argon gas.
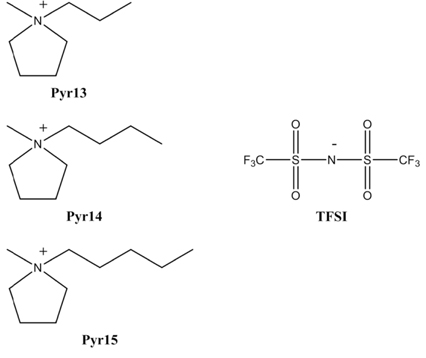
Chemical structure of the ionic liquids (Pyr13-TFSI, Pyr14-TFSI, Pyr15-TFSI) considered in this study.
2.2. Measurements
The conductivity measurements of Pyr-based ionic liquid electrolytes were performed using a Cond 3210 conductivity meter (WTW GmbH, Germany). The viscosity of the ionic liquid electrolytes was measured with a viscometer (uVISC, Rheosense). Cyclic voltammetry was performed using a Zahner Electrik IM6 over a voltage range of 0.1 to 2.7 V with various scan rates (10-200 mV s−1) at 55 ℃. Charge and discharge cycling tests of the cells were conducted at a constant current density of 3.54 mA cm−2 over a voltage range of 0.1 to 2.7 V at 25 and 55 ℃ using battery test equipment (PEBC 50.2, PNE solution). The morphology of the electrode surface after cycling was examined using a field emission scanning electron microscope (FE-SEM, JEOL JSM-6330F). Surface characterization of the carbon electrode in the cell after 1,000 cycles at 55 ℃ was performed using X-ray photoelectron spectroscopy (XPS). Prior to collecting the data, the electrodes were washed several times with anhydrous dimethyl carbonate to remove residual electrolyte, followed by vacuum drying overnight at room temperature. XPS measurements were conducted on a VG Multilab ESCA 2000 system using Al Kα radiation.
3. Results and Discussion
Fig. 2 presents the ionic conductivities and viscosities of the Pyr-based ionic liquid electrolytes considered in this study, which were measured at 25 and 55 ℃. The viscosity of Pyr13-TFSI was 55.6 cP at 25 ℃, which was lower than those of Pyr14-TFSI and Pyr15-TFSI. When the alkyl chain length in pyrrolidinium cation increased, the viscosity was increased due to higher molecular interaction. The ionic conductivities of the ionic liquid electrolytes decreased with increasing viscosity due to the reduction of ionic mobility. Accordingly, the Pyr13-TFSI exhibited the highest ionic conductivity among the ionic liquid electrolytes investigated. The ionic conductivities were increased and the viscosities were decreased as the temperature increased from 25 to 55 ℃, as shown in Fig. 2(b). At 55 ℃, the Pyr-based ionic liquid electrolytes exhibited ionic conductivities ranging from 1.0 to 1.7 mS cm−1.

Ionic conductivities and viscosities of Pyr-based ionic liquid electrolytes with various alkyl chain lengths, which were measured at (a) 25 ℃ and (b) 55 ℃.
The electrochemical properties of the EDLCs were investigated by cyclic voltammograms of the two-electrode cells. The cyclic voltammograms obtained at various scan rates and 55 ℃ in cells with different Pyr-based ionic liquid electrolytes are presented in Fig. 3. The cyclic voltammograms of all cells show rectangular shapes without faradaic current peaks at all scan rates, showing highly efficient capacitive behavior with good charge propagation. The shapes of the cyclic voltammograms obtained in various electrolytes were similar, irrespective of the type of ionic liquid electrolyte. However, it should be noted that the capacitive current increased with decreasing the alkyl chain lengths in the pyrrolidinium cation at the same scan rate, and thus the current was the highest in the cell assembled with Pyr13-TFSI when comparing current values at the same scan rate. This result can be ascribed to the higher ionic conductivity arising from lower viscosity of Pyr13-TFSI compared to those of other ionic liquid electrolytes, as explained in Fig. 2. The current was increased with the scan rate, as expected. The rectangular shape of the cyclic voltammogram was well maintained even at a scan rate as high as 200 mV s−1, which suggests that adsorption and desorption of both ions occur reversibly even at high rates in the electric double layer of the activated carbon powders.

Cyclic voltammograms obtained in the symmetric two-electrode cells assembled with various ionic liquid electrolytes, which were measured at various scan rates and at 55 ℃: (a) Pyr13-TFSI, (b) Pyr14-TFSI, and (c) Pyr15-TFSI.
The EDLC cells composed of activated carbon electrodes and various ionic liquid electrolytes were subject to galvanostatic charge-discharge cycling in a voltage range of 0.1-2.7 V at a constant current of 3.54 mA cm−2. Fig. 4(a) shows the voltage profiles of cells assembled with various ionic liquid electrolytes during the initial 5 cycles at 55 ℃. The charge-discharge curves exhibit a well-behaved triangular wave with no curvature and no large IR drop, indicating the prominent capacitive behavior of the cell. The specific capacitance of the electrode can be calculated using the equation C (Fg−1) = 2It / (mΔE), where I is the current applied for the charging and discharging, t is the time of discharge, ΔE is the voltage difference between the upper and lower potential limits, and m is the mass of the active materials (activated carbon) in one of the electrodes. The factor of 2 results from the total capacitance of the cell being measured over two equivalent single electrode capacitors in series [29,30]. By using the above equation, the initial specific capacitances of the cells assembled with Pyr13-TFSI, Pyr14-TFSI, and Pyr15-TFSI are calculated to be 131.6, 127.6, and 122.4 F g−1, respectively, based on activated carbon powder. The higher specific capacitance in the cell with Pyr13-TFSI can be attributed to the lower viscosity and higher ionic conductivity of Pyr13-TFSI, which is consistent with the cyclic voltammetry results discussed earlier. The capacitance retentions of the cells obtained from the galvanostatic charge-discharge cycles as a function of cycle number at 25 and 55 ℃ are presented in Fig. 4(b) and (c), respectively. As shown in Fig. 4(b), the cells exhibited very stable cycling behavior at 25 ℃. However, the capacitance retention was decreased with increasing temperature. After 1,000 cycles, the capacitance of the cell with Pyr13-TFSI retained 84.8 % of its initial capacitance at 55 ℃. The capacitance retention was improved with increasing alkyl chain length in the pyrrolidinium cation at 55 ℃; as a result, the cell with Pyr15-TFSI exhibited the best cycling stability with capacity retention of 91.1 % after 1,000 cycles. This result may arise from the differences in the reductive stability of Pyr-based ionic liquid electrolytes, because varying the pyrrolidinium cation has a profound impact on the reductive stability of the ionic liquid electrolyte. During the charge cycle, the potential of the negative electrode decreases, the electrolyte can be reductively decomposed on the electrode surface at low potentials, and the degradation products might adversely affect the cycling stability of the cells at high temperature.

(a) Charge and discharge curves of EDLCs assembled with various ionic liquid electrolytes during the initial five cycles at 55 ℃. Capacitance retention of EDLCs assembled with various ionic liquids as a function of the cycle number at (b) 25 ℃ and (c) 55 ℃, respectively (current density: 3.54 mA cm−2, cut-off voltage: 0.1-2.7 V).
In order to confirm the degradation products formed on the electrode surface due to the irreversible decomposition of the ionic liquid electrolyte, we obtained FE-SEM images of the negative electrode before and after 1,000 cycles in various ionic liquid electrolytes at 55 ℃. As shown in Fig. 5(a), the pristine electrode exhibited well distributed conducting carbon particles (super-P carbon) on the activated carbon powder. In contrast, the electrodes obtained after cycling in various ionic liquid electrolytes were covered with a relatively thick surface layer (Fig. 5(b) and 5(c)), which may be formed via the reductive decomposition of ionic liquid electrolytes on the electrode surface. These results provide conclusive evidence that the surface layer was formed via the irreversible decomposition of the ionic liquid electrolyte at the electrode surface during repeated cycling. When an electron is added to the pyrrolidinium cation, the resulting radical is decomposed into a neutral molecule and a smaller radical. According to a previous report [31], the reduction of the Pyr14 cation leads to the formation of methylpyrrolidine, butyl radical, dibutylmethylamine radical, butylpyrrolidine, and methyl radical through various decomposition mechanisms, as demonstrated in Fig. 6. These radicals can react to form a passive film on the electrode surface; consequently, the resistive surface film formed on the electrode hampers the electrochemical reaction at the electrode and electrolyte interface, which causes a decline in capacitance with cycling, as shown in Fig. 4(c).

FE-SEM images of the negative electrode surface before and after cycling in various ionic liquid electrolytes at 55 ℃: (a) pristine electrode, electrodes cycled in (b) Pyr13-TFSI, (c) Pyr14-TSFI, and (d) Pyr15-TFSI.

Proposed mechanism for reductive decomposition of the 1-methyl-1-butylpyrrolidinium (Pyr14) cation to (a) methylpyrrolidine and butyl radical, (b) dibutylmethylamine radical, and (c) butylpyrrolidine and methyl radical.
In order to examine the chemical compounds on the surface of the electrodes cycled in various ionic liquid electrolytes, XPS measurements were performed after 1,000 cycles, and the resulting C 1s XPS spectra are shown in Fig. 7. According to previous literature, the peak observed at 284.5 eV is attributed to the C-C bond in carbon, and the peak appearing at 292.0 eV corresponds to the C-F bond in the PTFE binder [32-34]. The C-N peak observed at 285.7 eV can be generated via the decomposition of the pyrrolidinium cation on the electrode surface [35]. This result suggests that the surface layer formed on the electrode surface after 1,000 cycles arises from the reductive decomposition of pyrrolidinium cations (Pyr13, Pyr14, Pyr15) during repeated cycling. It is noticeable that the intensity of the C-N peak decreased as the alkyl chain length increased in the pyrrolidinium cation. When the pyrrolidinium cation contains a longer alkyl chain, the cation is more resistive to reductive decomposition due to the structural hindrance of access to electrons. Thus, the intensity of the C-N peak arising from the reductive decomposition of the pyrrolidinium cation is the lowest in the electrode cycled in Pyr15-TFSI, indicating that the Pyr15 cation is the most stable against irreversible decomposition, resulting in improved cycling stability. From the above results, it is concluded that the longer alkyl chain in the Pyr cation improves the cycling stability of EDLCs by suppressing the reductive decomposition of ionic liquid electrolytes during cycling at high temperature.

C 1s XPS spectra of negative electrodes cycled in various ionic liquid electrolytes at 55 ℃: (a) Pyr13-TFSI, (b) Pyr14-TSFI and (c) Pyr15-TFSI.
4. Conclusion
Pyrrolidinium-based ionic liquids with various types of cations were used as electrolytes for high-temperature EDLCs. The cells assembled with pyrrolidinium-based ionic liquid electrolytes exhibited initial specific capacitances ranging from 122.4 to 131.6 F g−1 at 55 ℃. The initial specific capacitance decreased as the alkyl chain length in the pyrrolidinium cation increased due to the lower ionic conductivity and higher viscosity of the ionic liquid electrolyte. With respect to capacitance retention, the cell assembled with Pyr-TFSI containing a longer alkyl chain showed more stable cycling characteristics via suppression of the reductive decomposition of the pyrrolidinium cation during repeated cycling.
Acknowledgements
This work was supported by the Industrial Promotion Program of Economic Cooperation Area of MOTIE/KIAT [R0004005].
References
M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta., 2006, 51, 5567-5580.
Galinski M., Lewandowski A., Stepniak I.. Electrochim. Acta. 2006;51:5567–5580. 10.1016/j.electacta.2006.03.016.S. Pandey, Anal. Chim. Acta, 2006, 556, 38-45.
Pandey S.. Anal. Chim. Acta 2006;556:38–45. 10.1016/j.aca.2005.06.038.M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621-629.
Armand M., Endres F., MacFarlane D. R., Ohno H., Scrosati B.. Nat. Mater. 2009;8:621–629. 10.1038/nmat2448.W. A. Henderson and S. Passerini, Chem. Mater., 2004, 16, 2881-2885.
Henderson W. A., Passerini S.. Chem. Mater. 2004;16:2881–2885. 10.1021/cm049942j.Q. Zhou, P. D. Boyle, L. Malpezzi, A. Mele, J.-H. Shin, S. Passerini and W. A. Henderson, Chem. Mater., 2011, 23, 4331-4337.
Zhou Q., Boyle P. D., Malpezzi L., Mele A., Shin J.-H., Passerini S., Henderson W. A.. Chem. Mater. 2011;23:4331–4337. 10.1021/cm201427k.J. Vatamanu, Z. Hu, D. Bedrov, C. Perez and Y. Gogotsi, J. Phys. Chem. Lett., 2013, 4, 2829-2837.
Vatamanu J., Hu Z., Bedrov D., Perez C., Gogotsi Y.. J. Phys. Chem. Lett. 2013;4:2829–2837. 10.1021/jz401472c.D. R. MacFarlane, M. Forsyth, P. C. Howlett, J. M. Pringle, J. Sun, G. Annat, W. Neil and E. I. Izgorodina, Acc. Chem. Res., 2007, 40, 1165-1173.
MacFarlane D. R., Forsyth M., Howlett P. C., Pringle J. M., Sun J., Annat G., Neil W., Izgorodina E. I.. Acc. Chem. Res. 2007;40:1165–1173. 10.1021/ar7000952.S. R. Sivakkumar, D. R. MacFarlane, M. Forsyth and D. W. Kim, J. Electrochem. Soc., 2007, 154, A834-A838.
Sivakkumar S. R., MacFarlane D. R., Forsyth M., Kim D. W.. J. Electrochem. Soc. 2007;154:A834–A838.A. Lewandowski, A. Olejniczak, M. Galinski and I. Stepniak, J. Power Sources, 2010, 195, 5814-5819.
Lewandowski A., Olejniczak A., Galinski M., Stepniak I.. J. Power Sources 2010;195:5814–5819. 10.1016/j.jpowsour.2010.03.082.M. Mastragostino and F. Soavi, J. Power Sources, 2007, 174, 89-93.
Mastragostino M., Soavi F.. J. Power Sources 2007;174:89–93. 10.1016/j.jpowsour.2007.06.009.C. Arbizzani, S. Beninati, M. Lazzari, F. Soavi and M. Mastragostino, J. Power Sources, 2007, 174, 648-652.
Arbizzani C., Beninati S., Lazzari M., Soavi F., Mastragostino M.. J. Power Sources 2007;174:648–652. 10.1016/j.jpowsour.2007.06.162.C. Arbizzani, M. Biso, D. Cericola, M. Lazzari, F. Soavi and M. Mastragostino, J. Power Sources, 2008, 185, 1575-1579.
Arbizzani C., Biso M., Cericola D., Lazzari M., Soavi F., Mastragostino M.. J. Power Sources 2008;185:1575–1579. 10.1016/j.jpowsour.2008.09.016.A. Balducci, R. Dugas, P. L. Taberna, P. Simon, D. Plee, M. Mastragostino and S. Passerini, J. Power Sources, 2007, 165, 922-927.
Balducci A., Dugas R., Taberna P. L., Simon P., Plee D., Mastragostino M., Passerini S.. J. Power Sources 2007;165:922–927. 10.1016/j.jpowsour.2006.12.048.A. Lewandowski and A. Swiderska, Solid State Ionics, 2003, 161, 243-249.
Lewandowski A., Swiderska A.. Solid State Ionics 2003;161:243–249. 10.1016/S0167-2738(03)00275-3.L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520-2531.
Zhang L. L., Zhao X. S.. Chem. Soc. Rev. 2009;38:2520–2531. 10.1039/b813846j.B. Xu, F. Wu, R. Chen, G. Cao, S. Chen, G. Wang and Y. Yang, J. Power Sources, 2006, 158, 773-778.
Xu B., Wu F., Chen R., Cao G., Chen S., Wang G., Yang Y.. J. Power Sources 2006;158:773–778. 10.1016/j.jpowsour.2005.08.043.M. Ue, K. Ida and S. Mori, J. Electrochem. Soc., 1994, 141, 2989-2994.
Ue M., Ida K., Mori S.. J. Electrochem. Soc. 1994;141:2989–2994. 10.1149/1.2059270.T. Sato, G. Masuda and K. Takagi, Electrochim. Acta, 2004, 49, 3603-3611.
Sato T., Masuda G., Takagi K.. Electrochim. Acta 2004;49:3603–3611. 10.1016/j.electacta.2004.03.030.M. Ue, M. Takeda, A. Toriumi, A. Kominato, R. Hagiwara and Y. Ito, J. Electrochem. Soc., 2003, 150, A499-A502.
Ue M., Takeda M., Toriumi A., Kominato A., Hagiwara R., Ito Y.. J. Electrochem. Soc 2003;150:A499–A502. 10.1149/1.1559069.A. Lewandowski and M. Galinski, J. Phys. Chem. Solids, 2004, 65, 281-286.
Lewandowski A., Galinski M.. J. Phys. Chem. Solids 2004;65:281–286. 10.1016/j.jpcs.2003.09.009.J. D. Stenger-Smith, C. K. Webber, N. Anderson, A. P. Chafin, K. Zong and J. R. Reynolds, J. Electrochem. Soc., 2002, 149, A973-A977.
Stenger-Smith J. D., Webber C. K., Anderson N., Chafin A. P., Zong K., Reynolds J. R.. J. Electrochem. Soc. 2002;149:A973–A977. 10.1149/1.1485773.A. Balducci, W. A. Henderson, M. Mastragostino, S. Passerini, P. Simon and F. Soavi, Electrochim. Acta, 2005, 50, 2233-2237.
Balducci A., Henderson W. A., Mastragostino M., Passerini S., Simon P., Soavi F.. Electrochim. Acta 2005;50:2233–2237. 10.1016/j.electacta.2004.10.006.B. Xu, F. Wu, R. Chen, G. Cao, S. Chen, G. Wang and Y. Yang, J. Power Sources, 2006, 158, 773-778.
Xu B., Wu F., Chen R., Cao G., Chen S., Wang G., Yang Y.. J. Power Sources 2006;158:773–778. 10.1016/j.jpowsour.2005.08.043.J. N. Barisci, G. G. Wallace, D. R. MacFarlane and R. H. Baughman, Electrochem. Comm., 2004, 6, 22-27.
Barisci J. N., Wallace G. G., MacFarlane D. R., Baughman R. H.. Electrochem. Comm. 2004;6:22–27. 10.1016/j.elecom.2003.09.015.H. Kurig, M. Vestli, K. Tonurist, A. Janes and E. Lust, J. Electrochem. Soc., 2012, 159, A944-A951.
Kurig H., Vestli M., Tonurist K., Janes A., Lust E.. J. Electrochem. Soc. 2012;159:A944–A951. 10.1149/2.022207jes.A. Brandt, S. Pohlmann, A. Varzi, A. Balducci and S. Passerini, MRS Bulletin, 2013, 38, 554-559.
Brandt A., Pohlmann S., Varzi A., Balducci A., Passerini S.. MRS Bulletin 2013;38:554–559. 10.1557/mrs.2013.151.C. Largeot, P. L. Taberna, Y. Gogotsi and P. Simon, Electrochem. Solid-State Lett., 2011, 14, A174-A176.
Largeot C., Taberna P. L., Gogotsi Y., Simon P.. Electrochem. Solid-State Lett 2011;14:A174–A176. 10.1149/2.013112esl.M. Montanino, F. Alessandrini, S. Passerini and G. B. Appetecchi, Electrochim. Acta, 2013, 96, 124-133.
Montanino M., Alessandrini F., Passerini S., Appetecchi G. B.. Electrochim. Acta 2013;96:124–133. 10.1016/j.electacta.2013.02.082.S. Paul, J. H. Kim and D. W. Kim, J. Electrochem. Sci. and Tech., 2011, 2, 91-96.
Paul S., Kim J. H., Kim D. W.. J. Electrochem. Sci. and Tech. 2011;2:91–96. 10.5229/JECST.2011.2.2.091.V. Khomenko, E. Frackowiak and F. Beguin, Electrochim. Acta, 2005, 50, 2499-2506.
Khomenko V., Frackowiak E., Beguin F.. Electrochim. Acta 2005;50:2499–2506. 10.1016/j.electacta.2004.10.078.M. C. Kroon, W. Buijs, C. J. Peters and G.-J. Witkamp, Green Chem., 2006, 8, 241-245.
Kroon M. C., Buijs W., Peters C. J., Witkamp G.-J.. Green Chem. 2006;8:241–245. 10.1039/B512724F.Z. Yue, J. Economy and G. Bordson, J. Mater. Chem., 2006, 16, 1456-1461.
Yue Z., Economy J., Bordson G.. J. Mater. Chem. 2006;16:1456–1461. 10.1039/b513267c.M. Schulze, K. Bolwin, E. Giilzow and W. Schnurnberger, Fresenius. J. Anal. Chem., 1995, 353, 778-784.
Schulze M., Bolwin K., Giilzow E., Schnurnberger W.. Fresenius. J. Anal. Chem. 1995;353:778–784. 10.1007/BF00321370.A. M. Bittner, M. Zhu, Y. Yang, H. F. Waibel, M. Konuma, U. Starke and C. J. Weber, J. Power Sources, 2012, 203, 262-273.
Bittner A. M., Zhu M., Yang Y., Waibel H. F., Konuma M., Starke U., Weber C. J.. J. Power Sources 2012;203:262–273. 10.1016/j.jpowsour.2011.10.083.S. Men, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 15244-15255.
Men S., Lovelock K. R. J., Licence P.. Phys. Chem. Chem. Phys. 2011;13:15244–15255. 10.1039/c1cp21053j.