Amorphous Vanadium Titanates as a Negative Electrode for Lithium-ion Batteries
Article information
Abstract
Amorphous vanadium titanates (aVTOs) are examined for use as a negative electrode in lithium-ion batteries. These amorphous mixed oxides are synthesized in nanosized particles (<100 nm) and flocculated to form secondary particles. The V5+ ions in aVTO are found to occupy tetrahedral sites, whereas the Ti4+ ions show fivefold coordination. Both are uniformly dispersed at the atomic scale in the amorphous oxide matrix, which has abundant structural defects. The first reversible capacity of an aVTO electrode (295 mA h g−1) is larger than that observed for a physically mixed electrode (1:2 aV2O5 | aTiO2, 245 mA h g−1). The discrepancy seems to be due to the unique four-coordinated V5+ ions in aVTO, which either are more electron-accepting or generate more structural defects that serve as Li+ storage sites. Coin-type Li/aVTO cells show a large irreversible capacity in the first cycle. When they are prepared under nitrogen (aVTO-N), the population of surface hydroxyl groups is greatly reduced. These groups irreversibly produce highly resistive inorganic compounds (LiOH and Li2O), leading to increased irreversible capacity and electrode resistance. As a result, the material prepared under nitrogen shows higher Coulombic efficiency and rate capability.
1. Introduction
Since lithium-ion batteries (LIBs) were first commercialized in 1991, the market for their use in portable electronic devices and electric vehicles (EVs) has grown steadily [1,2]. The market could be further expanded if the cell performance is improved in terms of capacity, power, safety, and cycle life [3]. Unfortunately, current LIBs cannot fully satisfy the high-power demand of EVs. One reason for this limitation is the use of graphite negative electrodes; the working potential of graphite is so close to that of Li metal that Li plating is highly probable under high-rate charging and frequently leads to severe safety problems such as internal shorting by dendritic Li metal.
Ti-based oxides such as lithium titanate (Li4−Ti5O12, LTO) [4-7] and titanium oxides (TiO2) [8-10] have emerged as alternatives to graphite for negative electrode materials in high-power applications. Here, Li plating is unlikely even under high-current charging because their working potential (1.5-1.8 V vs. Li/Li+) is much higher than that of Li metal. The high-current performance of TiO2 is, however, largely offset by the limited specific capacity (<200 mA h g−1). Their latent specific capacity cannot be fully utilized because of irreversible changes to inactive phases upon deep charging [11]. To prevent or mitigate the irreversible phase transitions, TiO2 has been prepared in nanostructured forms or amorphous phases. It is intuitive that phase transitions can be suppressed by introducing disorder, for instance, structural defects such as vacancies and void spaces on the surface of nanostructured materials or in the bulk of amorphous materials [12]. Moreover, nanostructuring of electrode materials is expected to be a promising strategy to increase the power density of cells by shortening the Li+ diffusion path within the bulk materials [13-17]. In addition to suppressing unwanted phase transitions, amorphization of electrode materials has also been proven effective for increasing the number of Li+ storage sites and facilitating solid-state Li+ diffusion through the abundant void spaces [18-21]. The reason is very likely that the structural defects (vacancies and void spaces) can serve as Li+ storage sites, as is well-known for hard carbons [22,23]. Recent studies have demonstrated that several amorphous materials can be successfully applied in Na+ ion batteries because the amorphous framework provides Na+ storage sites and ion diffusion channels [24,25].
A literature survey reveals that the rate performance of TiO2 electrodes can be improved somewhat by amorphization, but their specific capacity cannot be increased [18,26,27]. The capacity limitation can be understood by considering that lithiation proceeds via co-injection of Li+ ions and the equivalent quantity of electrons. Electrode materials should carry both Li+ storage sites and electron storage sites (redox centers). Even if the number of Li+ storage sites (vacancies and void spaces) can be increased by amorphization of TiO2, the electron-accepting ability cannot be enlarged by amorphization because the redox centers are the metal (Ti) ions in TiO2. In this work, to increase the specific capacity of amorphous TiO2 (aTiO2), a mixed oxide containing Ti and V ions was prepared on the basis of information in previous reports. As reported in a previous study, V5+ ions in amorphous V2O5 (aV2O5) have better electron uptake than Ti4+ in aTiO2 at >1.0 V [28-31]. If this is the case in the mixed oxide prepared in this work, V5+ ions can show a higher electron-accepting ability as a redox center than Ti4+ ions; the net result will be an increase in the specific capacity. A further role of V5+ ions is that homogeneous mixing of V5+ and Ti4+ ions in the oxide matrix can produce a larger number of structural disorders that can act as Li+ storage sites [32,33].
As a way to introduce V5+ ions into aTiO2, amorphous vanadium titanate (aVTO) was synthesized using a simple precipitation method. The following features of the synthesized aVTO were examined: (i) whether the V and Ti ions in aVTO are homogeneously mixed at an atomic scale instead of existing in a physical mixture of aV2O5 and aTiO2, (ii) how the oxidation state of the V and Ti ions and the surrounding local structure in aVTO differ from those in their respective oxides (aV2O5 and aTiO2), and (iii) how the Li+ storage sites and redox centers in aVTO differ from those in their respective oxides. Another objective of this work was to decrease the quantity of surface impurities such as surface hydroxyl groups and residual water, which are known to cause side reactions that lower the Coulombic efficiency and increase the electrode resistance [20,34,35]. In this work, the amorphous materials were synthesized by a precipitation method without heat treatment; a high population of surface hydroxyl groups and residual water was unavoidable. The effects of these impurities on the electrode performance of aVTO were examined. To avoid these impurities, the synthetic atmosphere was changed from air to nitrogen, and the effect of this change on the impurity levels and electrochemical performance was also examined.
2. Experimental Section
Amorphous vanadium titanates (aVTOs) were synthesized by a precipitation method. Vanadium oxide sulfate hydrate (2.5 g, VOSO4·xH2O, Alfa Aesar) and titanium oxide sulfate sulfuric acid hydrate (2.5 g, TiOSO4·xH2O + H2SO4, Alfa Aesar) were dissolved in 250 mL of distilled water. Then, the precursor solution was dropped into a gas (air or nitrogen)-purged urea solution to obtain precipitates [36]. The reaction temperature was fixed at 60°C, and an ammonia solution was added dropwise to the solution to maintain the solution pH at 7. The precipitates obtained under air and nitrogen atmospheres are denoted aVTO and aVTO-N, respectively. The resulting precipitates were centrifuged and washed with distilled water; this was followed by drying at 80°C overnight and additional drying at 300°C under vacuum for 2 h. For comparison, amorphous vanadium pentoxide (aV2O5) and titanium oxide (aTiO2) were also synthesized using the same procedure.
The particle morphology was examined using field-emission scanning electron microscopy (FE-SEM, JEOL JSM-6700F). Energy-dispersive X-ray spectroscopy (EDS) mapping was performed using a scanning transmission electron microscope (STEM, Tecnai F20). The surface area was measured from the nitrogen adsorption isotherm using a Micromeritics ASAP 2010 instrument. The X-ray diffraction (XRD) patterns were obtained using a Bruker D8 diffractometer equipped with Cu Kα radiation (1.54056 Å). The atomic ratio of titanium and vanadium in aVTO was measured by the inductively coupled plasma (ICP) technique using the Optima-4300 DV installed at the National Center for Inter-University Research Facilities at Seoul National University. X-ray absorption spectroscopy data for the titanium K-edge (E0 = 4966 eV) and vanadium K-edge (E0 = 5465.1 eV) were obtained using transmission mode with a ring current of 320-400 mA at 3.0 GeV at the Pohang Light Source. A Si (111) monochromator crystal was used with detuning to 70% in intensity to eliminate the high-order harmonics. Energy calibration for the titanium and vanadium K-edges was conducted using titanium and vanadium metal foil. Fourier transform infrared (FTIR) transmission spectra for aVTO and the reference oxides were measured using a Bruker Vertex 80V IR spectrometer in vacuum. The samples for the transmission spectra were pelletized with potassium bromide (KBr).
To prepare the composite electrodes, the synthesized metal oxides, Super-P, styrene-butadiene rubber (SBR), and carboxymethyl cellulose (CMC) (80:10:5:5 in weight ratio) were mixed in an agate mortar and dispersed in distilled water. The slurry was then coated on copper foil (LS wire, thickness 20 μm) with a doctor blade in a drying room. The composite electrodes were punched and dried at 120°C overnight in vacuum. The mass loading of active material in the electrodes was 1.6-2.0 mg cm−2. Coin cells (2032-type) were fabricated in an argon-filled dry box. Lithium foil and a trilayered polypropylene-polyethylene membrane (PP-PE-PP, Celgard 2320) were used as the counter electrode and separator, respectively. LiPF6 (1.0 M) dissolved in ethylene carbonate and dimethyl carbonate (DMC) (1:2 in volume ratio) was used as the electrolyte. Galvanostatic charge/discharge measurements were performed at a current density of 100 mA g−1 over the potential range of 0.8-3.0 V (vs. Li/Li+). X-ray photoelectron spectroscopy (XPS) data were collected in an ultrahigh-vacuum multipurpose surface analysis system (Sigma Probe, Thermo Scientific, UK) operating at a base pressure of <10−10 mbar. The photoelectrons were excited by an Al Kα (1486.6 eV) anode operating at a constant power of 100 W (15 kV and 10 mA). For ex situ XPS analysis, aVTO and aVTO-N electrodes were prepared by embedding the powder in a copper foil to eliminate interference coming from the conductive carbon (Super-P) and the polymer binder (SBR-CMC). The cycled cells were disassembled, and the electrode samples were washed with DMC in an argon-filled glove box. They were then transferred to the XPS instrument without air exposure. The binding energy was calibrated from the hydrocarbon contamination using the C 1s peak at 285.0 eV.
3. Results and Discussion
Fig. 1a and 1b show FE-SEM images of the synthesized aVTO powder. Its size was approximately several microns, and it consisted of small primary particles <100 nm in size, resulting in a large surface area of 54.39 m2 g−1. The morphology of aVTO powder is advantageous in high-power applications because it provides a short Li+ diffusion path and a larger contact area with the electrolyte solution [13-17]. Fig. 1c-e show the STEM image and EDS mappings, which demonstrate that titanium and vanadium are homogeneously dispersed within the nanosized primary particles. The ICP measurement reveals that the atomic ratio of titanium and vanadium is close to 1:1. The XRD pattern obtained from the aVTO powder was featureless, indicating the amorphous nature of the synthesized powder (Fig. 1f).

(a) and (b) FE-SEM images of aVTO, (c) STEM image of aVTO, (d) and (e) EDS mappings of vanadium and titanium, and (f) XRD patterns of aVTO powder and aV2O5 + aTiO2 (1:2 in mole ratio) powder dried at 300°C under vacuum.
The metal valence and local structure of metal ions in aVTO, aV2O5, and aTiO2 were examined using X-ray absorption near-edge structure (XANES) spectra (Fig. 2a and 2b) and extended X-ray absorption fine structure (EXAFS) spectra (Fig. 2c and 2d). The appearance of the main edge of vanadium, which is a common feature of crystalline vanadium pentoxide (cV2O5) [37], indicates that the vanadium valence is 5+ for both aVTO and aV2O5 (Fig. 2a). The titanium valence is 4+ in both aVTO and aTiO2 because the main edge appears as it does for crystalline titanium oxide (cTiO2), which also has a Ti valence of 4+. The local structure of V5+ in aVTO and aV2O5 was analyzed using the data shown in Fig. 2a and 2c. The pre-edge of the vanadium K-edge does not appear if the local symmetry of V5+ is octahedral. With a decrease in the local symmetry from an octahedral geometry, the pre-edge of the vanadium K-edge becomes stronger owing to a formally forbidden dipole transition from the vanadium 1s orbital to oxygen 2p states hybridized with 3d orbitals [22]. As shown in Fig. 2a, the orthorhombic V2O5 (cV2O5) shows a strong pre-edge because vanadium ions are located at the five-coordinate (VO5) square pyramidal sites [29], which exhibit much lower symmetry than the six-coordinate octahedral structure. This strong pre-edge peak is also observed in both aV2O5 and aVTO, implying that the local structure of V5+ is not octahedral. The detailed local structure can be estimated from the V-O shell depicted in Fig. 2c. The V-O peak of orthorhombic V2O5 (cV2O5) has low intensity and is divided into two (black line), which must be due to the fact that the V-O shell is composed of one short V = O bond (1.07 Å) and four long V-O bonds (1.53 Å) (five-coordinate square pyramidal sites, as shown in Fig. 2a). aV2O5 also shows a similar V-O shell comprising a short V = O bond and long V-O bonds, although the former is more intense than the latter (blue line), illustrating that the local structure of V5+ in aV2O5 does not differ greatly from that in cV2O5 (five-coordinate). However, the local structure of V5+ in aVTO is not five-coordinate, as demonstrated by the EXAFS data in Fig. 2c. Namely, the V-O shell produces an intensified single peak (red) at 1.20 Å, which corresponds to a tetrahedral structure around the V5+ ions [38]. The difference in V5+ local structure between aVTO (four-coordinate) and aV2O5 (five-coordinate) indicates that aVTO is not a physical mixture of aV2O5 and aTiO2; rather, the V5+ and Ti4+ ions are homogeneously mixed at the atomic scale. The local structure modification and homogeneous atomic scale mixing of V5+ can also be observed in the FTIR spectrum of aVTO (Fig. 2e). A sharp peak at about 995-1028 cm−1, indicative of a V = O bond, was significantly reduced. A broad shoulder between 950 and 980 cm−1, which is known to indicate the Ti-O-V bond, strongly implies chemical interaction between V5+ and Ti4+ [39].

X-ray absorption near-edge structures (XANES) data for aVTO and some reference oxides: (a); vanadium K-edge and (b); titanium K-edge. XANES data for amorphous V2O5 and TiO2 are taken from a physically mixed aTiO2 + aV2O5 electrode. Fourier transformed extended X-ray adsorption fine structures (EXAFS): (c); vanadium K-edge and (d); titanium K-edge for aVTO and reference oxides. EXAFS data for amorphous V2O5 and TiO2 are taken from a physically mixed aTiO2 + aV2O5 electrode and (e); Fourier Transform Infrared spectra (FTIR) measured in vacuum for KBr-pelletized aVTO, cV2O5, cTiO2 and a physical mixture of aV2O5 and aTiO2.
The local structure of Ti4+ in aVTO and aTiO2 was also analyzed. The titanium pre-edge peaks from crystalline (anatase) TiO2 (cTiO2) appear at 4969.2, 4970.8, 4972.0, and 4974.4 eV and are labeled A1, A2, A3, and B, respectively, as shown in the inset of Fig. 2b [40]. The first three (A1, A2, and A3) are due to dipole transitions to titanium 4p states hybridized with titanium 3d orbitals that are split into the t2g and eg bands, respectively [40]. As shown in Fig. 2b, the A2 and A3 peaks of aTiO2 and aVTO are much stronger than those of cTiO2, implying that the regular TiO6 octahedron, which is dominant in anatase TiO2, is distorted to form an irregular fivefold coordination (TiO5) structure in both aTiO2 and aVTO [18,41,42]. This feature can be ascertained from the radial distribution obtained from the Fourier transformation of the EXAFS spectra shown in Fig. 2d. The Ti–O bond in cTiO2 is located at 1.41 Å, but this peak is shifted to 1.35 Å for aTiO2 and aVTO. This observation agrees with the XANES spectra, which suggest irregular fivefold coordination of Ti4+ in aTiO2 and aVTO (Fig. 2b). That is, the Ti-O bond length is shorter for five-coordinate structures (aTiO2 and aVTO) than for the six-coordinate structure (cTiO2). In short, it is found that aVTO is composed of homogeneous mixing of irregular TiO5 and tetrahedral VO4 at the atomic scale rather than a physical mixture of the two separate oxides.
The galvanostatic lithiation and delithiation voltage profiles of the Li/aVTO cell are shown in Fig. 3a. The aVTO electrode shows a sloping voltage profile, which is a characteristic feature of amorphous electrodes, and exhibits a reversible capacity of 295 mA h g−1 in the first cycle. Note that this value is much larger than those of LTO (175 mA h g−1), anatase TiO2 (168 mA h g−1), and even amorphous TiO2, which shows a slightly larger capacity than anatase TiO2 (Fig. 3d) [7,43]. The electrode consisting of mechanically mixed aV2O5 and aTiO2, which were mixed in a 1:2 mole ratio to simulate the atomic ratio (1:1) in aVTO, delivers a reversible capacity of 245 mA h g−1 in the first cycle (Fig. 3c). Note that the reversible capacity of the aVTO electrode is larger than that of the mixed electrode, and the voltage profiles of the two are different in the first lithiation.

(a) Lithiation/delithiation voltage profile obtained from Li/aVTO cell, (b) differential capacity plot derived from (a), (c) lithiation/delithiation voltage profile obtained from Li/aV2O5 + aTiO2 (physical mixture in 1:2 mole ratio) cell, and (d) cycle performance of Li/aVTO, Li/aTiO2, and Li/aV2O5 + aTiO2 cells.
The redox behaviors of the two redox centers (V and Ti) in the aVTO and aV2O5 + aTiO2 electrodes were compared using ex situ XANES spectra (Fig. 4a and 4b). To obtain the relationship between the metal valence and absorption energy, XANES data were obtained from some reference oxides, namely, crystalline titanium oxides (cTiO2 and cTi2O3) and vanadium oxides (cV2O5, cVO2, and cV2O3), the metal valence of which has a formal value (for example, 4+ for cTiO2 and cVO2). The valence of the transition metal ions was calculated assuming that the metal valence is linearly related to the position of the main absorption edge. The electron energy at the half-height of the main absorption edge was taken and plotted, along with the estimated metal valences of the reference oxides. Note that the shape and position of the main edge depend on not only the metal valence but also the local structure and coordination of metal ions, such that the calculated values carry an appreciable uncertainty. As shown in Fig. 4c, the Ti valence in aVTO changes from 3.58+ to 3.74+. A comparable change is observed for aTiO2 (from 3.61+ to 3.81+), illustrating that the electron-accepting ability of Ti ions is comparable for aVTO and aTiO2. The redox behavior of V ions in aVTO and aV2O5 was also examined. As shown in Fig. 4d, the V valence in aVTO changes from 2.7+ to 4.6+, whereas it changes from 3.6+ to 4.7+ in aV2O5. Clearly, the oxidation state of V ions in aVTO is lower (2.7+) that that in aV2O5 (3.6+) upon lithiation, illustrating that the electron-accepting ability of V5+ ions as a redox center is greater in aVTO. As shown in Fig. 3a and 3c, the first reversible specific capacity of aVTO (295 mA h g−1) is larger than that of the physically mixed oxide (aV2O5 + aTiO2) (245 mA h g−1). Hence, the extra capacity (ca. 50 mA h g−1) delivered by aVTO can be attributed to the higher electron-accepting ability of the V5+ ions in aVTO. The apparent difference between aVTO and aV2O5 is due to the local structure; the V5+ ions in aVTO are four-coordinate, but those in aV2O5 are five-coordinate. The experimental data suggest that the difference in the local structure of the V5+ ions can cause different redox behavior.

Ex situ XANES spectra of (a) Ti K-edge and (b) V K-edge for aVTO, aTiO2, and aV2O5 electrodes, which were obtained in the fully lithiated state (0.8 V vs. Li/Li+) and fully delithiated state (3.0 V) in the first cycle. Metal valence changes for (c) Ti and (d) V upon the first lithiation, which were estimated from (a) and (b), respectively.
The extra capacity delivered by aVTO can also be explained in another way. Namely, aVTO should carry both Li+ storage sites and redox centers for lithiation. Two possibilities exist. First, if aVTO has abundant Li+ storage sites (structural defects), which is likely because it is amorphous, the number of redox centers (number of electrons to be stored) determines the overall Li storage capacity. If this is the case, the changes in the electronic structure of aVTO, which can be caused by modification of the local geometry by homogeneous mixing of cations, can change the Li storage capacity. One example is the Co3+ ions in the spinel-structured LiMn2-xCoxO4 and layered LiCoO2. Both the redox potential and redox capacity of Co3+ ions are different in the two oxides [44]. Considering that Ti substitution in V2O5 can alter the electronic structure of its original framework [45,46], it is very likely that the V ions in the V-Ti-O structure have a different redox behavior from those in V2O5. However, because it is difficult to analyze the band structure of amorphous materials, it is still unclear why the V ions in aVTO show a higher electron-accepting ability. The second possibility is the reverse case. Here, if the number of Li+ storage sites is smaller than that of redox centers, the overall capacity is determined by the former. If this is the case in aVTO, the larger specific capacity of aVTO over the mixed electrode (aV2O5 + aTiO2) can be accounted for by the generation of extra Li+ storage sites (structural defects) resulting from the addition of V5+ ions to the aTiO2 matrix. This possibility has been proposed in previous studies [32,33].
As shown in Fig. 3a and 3b, the Li/aVTO cell shows a large irreversible capacity in the first cycle, which diminishes in subsequent cycles. The major irreversible reactions occur at <2.0 V, as indicated by an arrow in Fig. 3b. Two possibilities exist for the irreversible reactions: electrolyte decomposition and Li reaction with residual water or surface hydroxyl groups. The former possibility is discarded because electrolyte decomposition is commonly observed at 0.7-0.8 V (vs. Li/Li+). The latter possibility seems more probable because the amorphous samples can carry residual water and surface hydroxyl groups, as they were prepared by drying at 300°C under vacuum. Two irreversible reactions (Eqns. 1 and 2) can be assumed [34,35]:
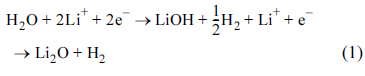

This is validated by the XPS spectra obtained from the aVTO electrode before and after lithiation (Fig. 5a and 5b). The O 1s spectra obtained before lithiation were deconvoluted into three peaks. The peak at 530.7 eV corresponds to the bulk oxygen in amorphous metal oxides. The O 1s photoelectrons at 532.7 and 534.0 eV come from the oxygen in hydroxyl groups and adsorbed water on the metal oxide surface, respectively [47]. As shown in Fig. 5a, the surface of aVTO is covered with hydroxyl groups. The four peaks in the O 1s spectra obtained after lithiation were assigned according to the reported binding energies: Li2O at 528.5 eV, LiOH at 531.5 eV, Li2CO3 and oxygen atoms doubly bound to carbon atoms at 532 eV, and oxygen bound to carbon with a single bond at 533.5 eV [48,49]. A large amount of Li2O and LiOH is found on the surface of lithiated aVTO. Clearly, the surface hydroxyl groups and residual water in aVTO are converted into Li2O and LiOH according to Eqns. 1 and 2, which appear as the irreversible capacity in Fig. 3a and 3b.

O 1s XPS spectra: (a) aVTO in the initial open-circuit voltage (OCV) state, (b) aVTO after lithiation down to 0.8 V, (c) aVTO-N in the initial OCV state, and (d) aVTO-N after lithiation down to 0.8 V.
Fig. 5c displays the O 1s XPS spectra obtained from the pristine aVTO-N electrode. The major O 1s photoelectrons are emitted from the lattice (bulk) oxygen. The population of surface hydroxyl groups and adsorbed water is much smaller than that in aVTO. The only difference between the two samples is the synthetic atmosphere (air and nitrogen). It is curious that more surface hydroxyl groups form in the presence of excess oxygen. The following reaction is proposed. Molecular oxygen can be converted into lattice oxygen, accompanied by generation of metal vacancies and holes as described by Eqn. 3. In the presence of water, the lattice oxygen and holes react further with water to generate surface hydroxyl groups as shown in Eqn. 4 [50].
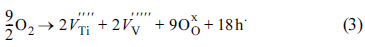
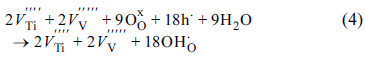
If this is the case, the reduced population of surface hydroxyl groups on aVTO-N can be rationalized.
The morphology and bulk properties of aVTO-N are very similar to those of aVTO even though aVTO-N has a larger surface area (62.77 m2 g−1). The metal valence is not changed even when the reaction atmosphere is changed from air to nitrogen. As shown in Fig. 6a, the electrode performance of the Li/aVTO-N cell, including the reversible capacity and cycling ability, is also comparable to that of Li/aVTO. The notable difference is the irreversible capacity in the first cycle. The irreversible capacity observed at <2.0 V in the aVTO electrode (Fig. 3b) is notably diminished in the aVTO-N electrode (Fig. 6b). As a result, the initial Coulombic efficiency increases in the Li/aVTO-N cell, as shown in Fig. 6c. Furthermore, the Li/aVTO-N cell shows higher Coulombic efficiency during cycling, indicating that the decreased quantity of surface hydroxyl groups positively affects the side reactions even after the first cycle.

(a) Lithiation/delithiation voltage profile obtained from the Li/aVTO-N cell, (b) differential capacity plot derived from (a), (c) comparison of the cycle data and Coulombic efficiency of aVTO and aVTO-N, and (d) rate performance of aVTO and aVTO-N electrodes.
The Li/aVTO-N cell outperforms the Li/aVTO cell with respect to the rate capability (Fig. 6d). As pointed out above, the only difference between the two electrodes is the quantity of surface hydroxyl groups; thus, the difference in rate capability should be explained on the basis of the quantity of surface hydroxyl groups. As shown in Fig. 5d, the quantity of inorganic components such as Li2O and LiOH, which are known to impede Li+ diffusion [51], is greatly diminished on the aVTO-N electrode. This must be due to the lower quantity of surface hydroxyl groups on aVTO-N. As a result, the formation of highly resistive inorganic compounds (Li2O and LiOH) is greatly suppressed during cell cycling. In short, a lower population of surface hydroxyl groups on aVTO-N is responsible for the decrease in irreversible capacity and the enhanced rate capability of the Li/aVTO-N cell.
4. Conclusions
To increase the specific capacity of amorphous titanium dioxide (aTiO2) electrodes, which are known to exhibit a reasonable rate capability, V5+ ions were incorporated into the aTiO2 matrix. The physicochemical properties and electrode performance of the resulting amorphous vanadium titanates (aVTOs) are summarized as follows.
(i) The prepared samples were not a physical mixture of aV2O5 and aTiO2. The V5+ and Ti4+ ions were homogeneously dispersed in the amorphous oxide matrix at the atomic scale.
(ii) The V5+ ions in aVTO were at four-coordinate sites, in contrast to the five-coordinate V5+ ions in aV2O5. However, fivefold coordination of Ti4+ ions appeared for aTiO2 and aVTO.
(iii) The oxide electrodes showed a sloping charge/discharge voltage profile, which is a characteristic feature of amorphous electrodes. Li+ storage in structural defects (vacancies, void spaces, cluster gaps, or interstitial sites) can thus be assumed.
(iv) The reversible specific capacity of aVTO was larger than that of the physically mixed (aV2O5 and aTiO2 in stoichiometric ratio) electrode. The extra capacity delivered by aVTO was thus attributed to contributions from the fourfold-coordinated V5+ ions in aVTO. Two possibilities were proposed for this contribution. One is the modification of the electronic structure by a local geometric change of V ions in aVTO. The other is the creation of extra Li+ storage sites (structural defects) by the incorporation of V5+ ions into the aTiO2 matrix.
(v) The surface hydroxyl groups and residual water in aVTO caused a serious irreversible reaction and formation of highly resistive inorganic species. The impurity population was, however, greatly reduced by changing the synthetic atmosphere from air to nitrogen. The amorphous vanadium titanate prepared under nitrogen (aVTO-N) outperformed that prepared under air in terms of the Coulombic efficiency and rate capability.
Acknowledgements
We acknowledge financial support from the National Research Foundation of Korea funded by the Ministry of Education, Science & Technology (MEST) (Grant NRF-2010- C1AAA001-2010-0029065). We thank the staff of the Research Institute of Advanced Materials (RIAM) for assistance with the STEM and EDS experiments.