


1. Introduction
During recent years, iron and cobalt alloy thin films, e.g. Fe-Ni, Fe-Pd and Co-Pd, have been widely used as the high density magnetic recording materials and magnetic sensors due to their remarkable perpendicular magnetic anisotropy [1-6]. Among these thin films, Fe70Pd30 film has a reversible thermoelastic austenite-martensite phase transformation [7,8], which makes it as an appropriate choice for application in microsensors, microactuators and micromagnets [9-11]. Another point that should be mentioned is the ferromagnetic property of both Fe-Pd martensite and austenite phases [11,12], which makes it distinctive among shape memory alloys as a ferromagnetic shape memory alloy (FSMA) [12].
Various synthesis methods have been employed for production of these alloys; including high vacuum sputtering [13], pulsed laser deposition [14], epitaxial vapor deposition [8], floating zone [15] and electrodeposition [4-7,16-22]. Not only electrodeposition process is favorable from the economical point of view, but also it brings the possibility of controlling chemical composition [7,16,20]. In addition, the response rate of sensors can be improved by the use of electrodeposition technique due to appropriate control on the structure size [7,12,23].
The choice of substrate material depends on the application of Fe-Pd films; e.g. for thermal sensors, copper and brass can be used due to their high thermal conduction. However, copper is a diamagnetic material [24] and in the case of magnetic incitement, brass is more suitable [2,20].
On the other hand, for examination the magneto-resistance (MR) property of Fe-Pd alloy films [25], the substrate should have higher electrical resistance in compared to the film [26]. Thus, semiconductor materials such as indium doped tin oxide (ITO) [26-28] or fluorine doped tin oxide (FTO) [29] can be employed as the substrate.
A literature survey shows that various substrates have been employed in Fe-Pd alloy electrodeposition including titanium [17], copper [18], platinum [19], brass [20], gold [21], and stainless steel [20]. However, less attention has been paid to the effects of substrate on the kinetics of electrodeposited films from electrochemical point of view [16-21]. For instance, Hernandez et al. evaluated the effect of different substrates (Au/Si, brass and stainless steel) on the chemical composition, magnetic properties and morphology of Fe-Pd alloy films [20]. But, there is no explanation about the substrate effects on the electrodeposition mechanisms.
Based on the above mentioned points, the main goal of present study is to investigate the effects of the substrate on the electrodeposition kinetics and consequently the structure of the electrodeposited Fe-Pd alloy films. Three different materials (copper, brass and FTO/glass) are employed as the substrates and Fe-Pd alloy are potentio-statically electrodeposited on them. The nucleation and growth kinetics, chemical composition as well as the cathodic current efficiency are identified and discussed from electrochemical points of view.
2. Experimental
2.1 Materials
The solutions were prepared by dissolving analytical grades of 200 mM FeSO4.7H2O and 20 mM PdCl2 salts in double distilled water. Also 0.6 M (NH4)2SO4 , 0.3 M Na3C6H5O7 and 0.25 M H3BO3 were employed as the complexing agents and supporting electrolyte. NH4(OH) was used to bind with palladium ions and also pH adjustment (pH = 8). Electrochemical experiments were conducted in a typical three-electrode cell. Copper and brass plates as well as sputtered FTO on glass were applied as the working electrodes. In Table 1 the properties of employed substrates are given. A platinum plate was placed at fixed distance (2 cm) in front of the working electrode and potentials were recorded versus saturated calomel reference electrode (SCE).
2.2 Analysis
Fe-Pd films were electrodeposited in the potential range of ŌłÆ850 to ŌłÆ1050 mV vs. SCE using a potentiostat/galvanostat, Autolab┬« model PGSTAT 302N connected to a computer (equipped by Nova 1.5 software). Chemical composition of alloy films was analyzed by energy dispersive X-ray spectroscopy (EDS) (Oxford Instrument 7538). The cathodic current efficiency (CCE) of the electrodeposition process was determined by measuring the weight of deposits and calculating the area under current density-time curves as following:
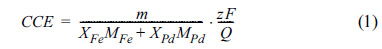
where m is the weight of deposit; XFe, XPd are the mole fractions of the alloy obtained from EDS analysis; MFe = 55.85, MPd = 106.4 gr molŌłÆ1 and Q is the surface area under j-t current transient curves for each cathodic potential (Coulomb).
Crystallographic structures were identified by the use of a Philips X-ray diffraction analysis with Cu K╬▒ radiation. Surface morphology of the alloy films were studied by atomic force microscopy (AFM) using a Digital Instruments Inc. Nano scope II and also by field emission scanning electron microscopy (FESEM), using a HIT-S4160.
3. Results and Discussion
3.1. Effect of substrate on chemical composition and cathodic current efficiency
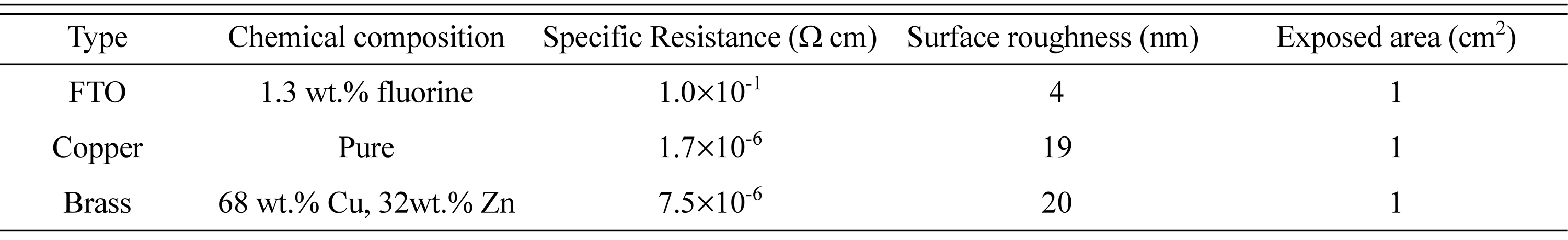
According to table of standard potentials in aqueous solution, there is a significant difference between redox potential of Fe2+(0.2 M)/Fe (ŌłÆ0.70 V vs. SCE) and Pd2+(0.02 M)/Pd (+0.66 V vs. SCE) [30,31]. However, formation of [Pd(NH3)4]2+ complex shifts the reversible potential of Pd to ŌłÆ0.24 V vs. SCE (based on constant stability of [Pd(NH3)4]2+; ╬▓4=1030.5) [32]. Fig. 1(a) shows the linear sweep voltammetry (LSV) curves corresponding to Cu, brass and FTO/glass electrodes. In the case of Cu and brass, two peaks appear in voltammograms which are associated to the Fe and Pd reduction [16]. The first peak in voltammograms is related to palladium diffusion-controlled region and next one to iron [33-36]. It is also found that for same overpotentials (Fig. 1), current density and accordingly deposition rate on the metallic substrates is higher than that of FTO due to the higher surface electrical resistance of FTO in compared to copper and brass (Table 1) [26]. Fig. 1(b) displays the LSV curves based on Fig. 1a. From Fig. 1(b), it is clear that there is a significant difference among the exchange current density (ECD) on copper, brass and FTO substrates. The various ECD may be ascribed to the under potential deposition (UPD) of Fe on copper and brass [16].
Fig.┬Ā1.
LSV curves during electrodeposition of Fe-Pd on different substrates, SR = 20 (mV SŌłÆ1), (a) linear current density and (b) logarithmic current density.
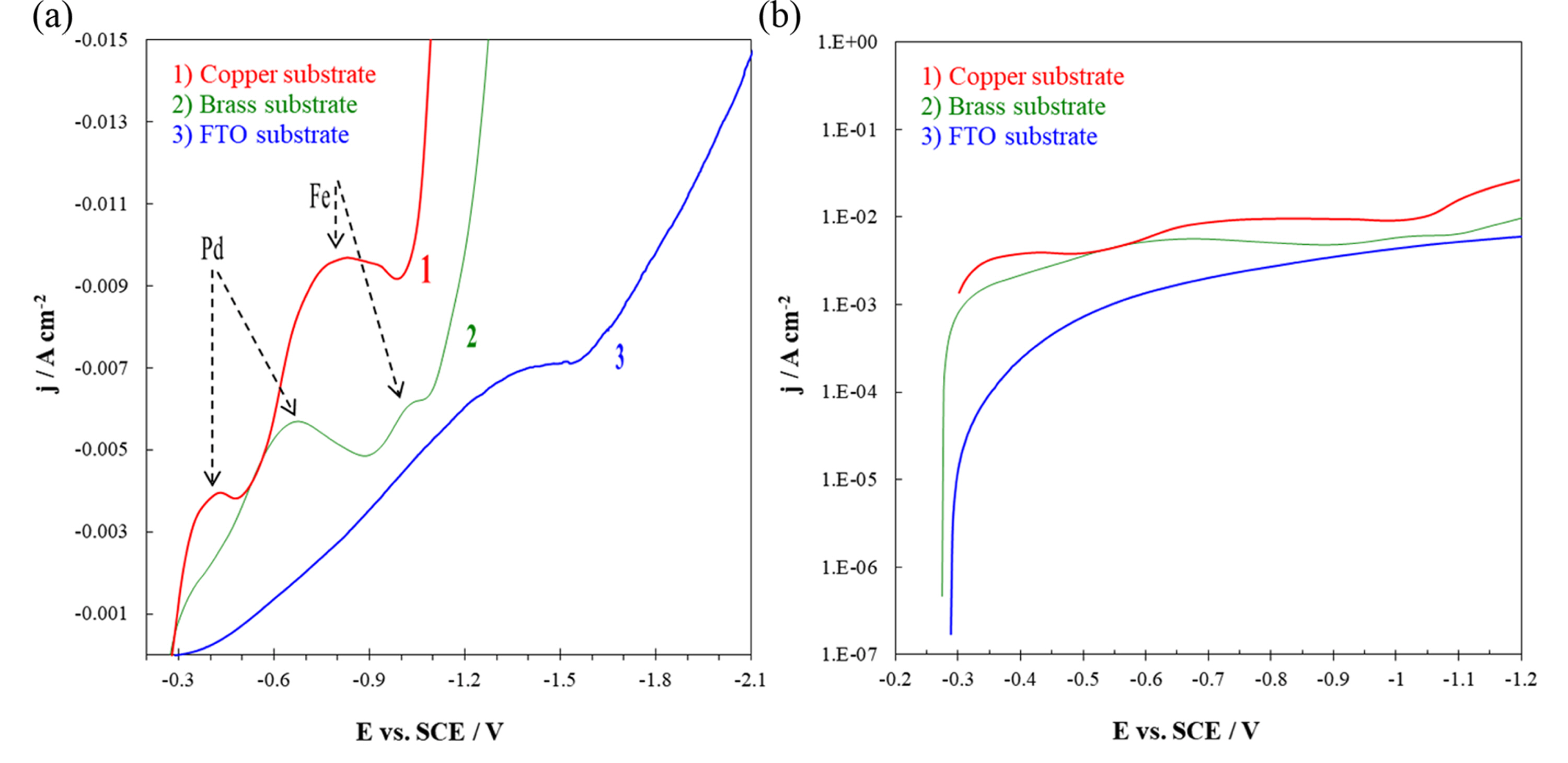
Fig. 2 (a-c) shows EDS spectra of the alloy films deposited on copper, brass and FTO substrates. The presence of peaks corresponding to iron and palladium confirm the co-deposition of Fe and Pd. Fig. 2(d) illustrates the chemical composition of the alloy films versus cathodic potential of electrodeposition. For different substrates, Fe content increases with an increase in overpotential (in the range of ca. 30 to 90 at.%). This anomalous co-deposition, i.e. preferential deposition of the less noble metal [37,38], has been previously reported for Fe-Pd alloy electrodeposition [16-18,21].
Fig.┬Ā2.
EDS spectra of Fe-Pd alloy film on (a) Copper, (b) Brass and (c) FTO/glass substrates. (d) Fe content of the thin films versus cathodic potential for electrodeposition of Fe-Pd alloy on different substrates; deposition time = 15 min.
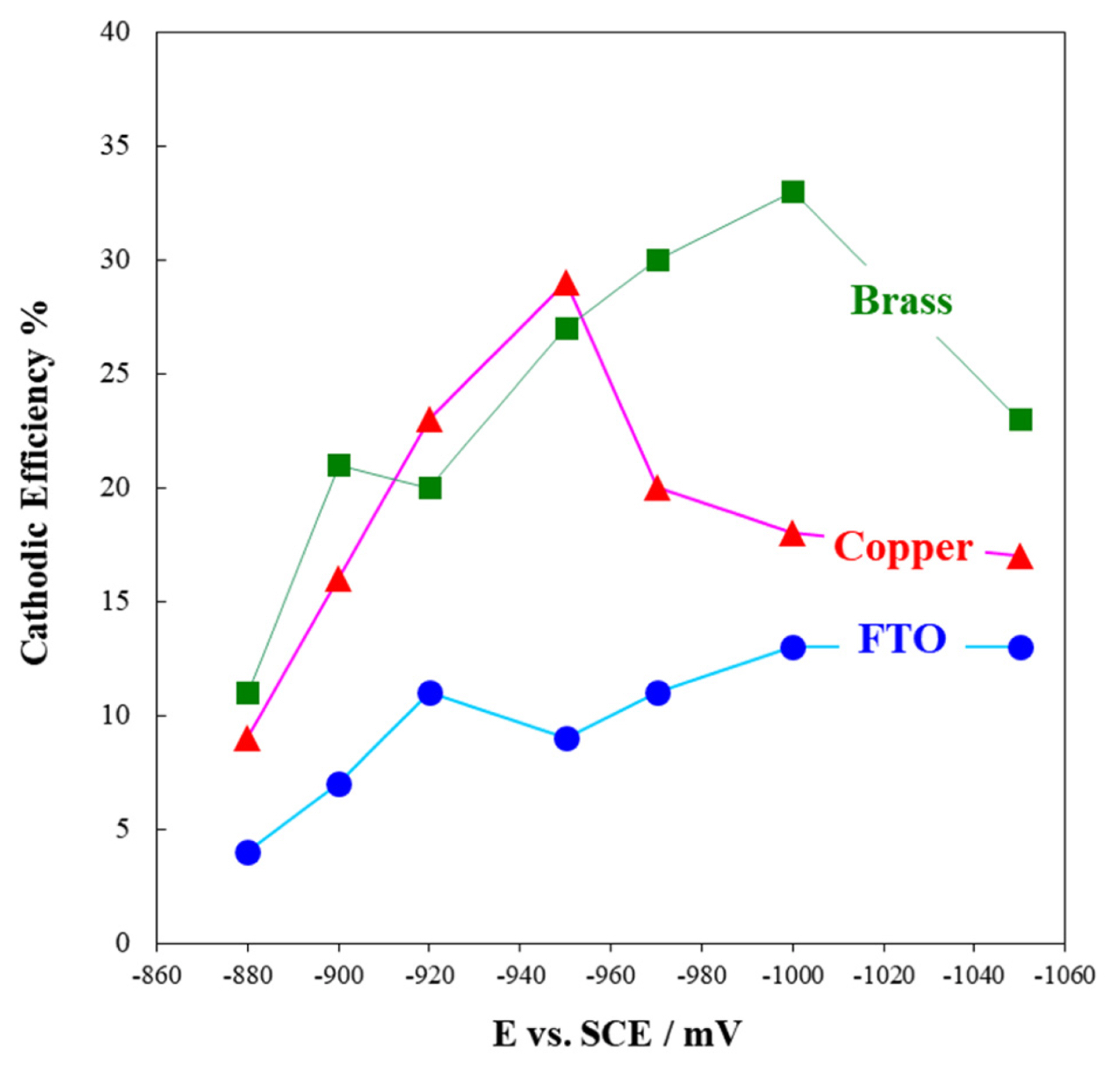
Fig. 3(a) illustrates the effect of overpotential on cathodic current efficiency (CCE). As seen, CCE is between 4 to 34% for different substrates. By considering the catalytic properties of Pd for H2 evolution as well as its hydrogen absorption property, the rate of hydrogen reduction on Pd nuclei during electrode-position is high and consequently, the CCE should be lower for Pd rich films due to higher hydrogen evolution rate. CCE-potential curves (Fig. 3(a)) corresponding to Cu and brass substrates, reveal that at low overpotentials, by a shift in potential to more cathodic values (and accordingly lower Pd content, Fig. 2(d)), CCE is enhanced. However at the potentials more cathodic than water decomposition potential (E = ŌłÆ950 to ŌłÆ1000 mV in Fig. 1), H2 evolution rate increases dramatically which cause to a sharp drop in CCE on these two substrates [39]. The same trend can be observed for FTO but there is no sharp drop in CCE because water decomposition potential on FTO substrate shifts to the more cathodic potentials (about ŌłÆ1.6 V, Fig. 1).
3.2. Effect of substrate on nucleation and growth
Fig. 4 displays SEM images of Fe-Pd deposits morphology on brass, copper and FTO substrates. The micrographs show an ultra-fine-grained structure in the films, and the average grain size corresponding to the deposits on Cu and brass is much smaller compared to FTO. The difference between grain sizes can be attributed to the different substrates roughness (Table 1).
Fig.┬Ā4.
FESEM images of Fe-Pd electrodeposited films at constant potential ŌłÆ950 mV vs. SCE. (a) Brass, (b) Copper and (c) FTO substrates. The scale bar represents 1 ╬╝m.

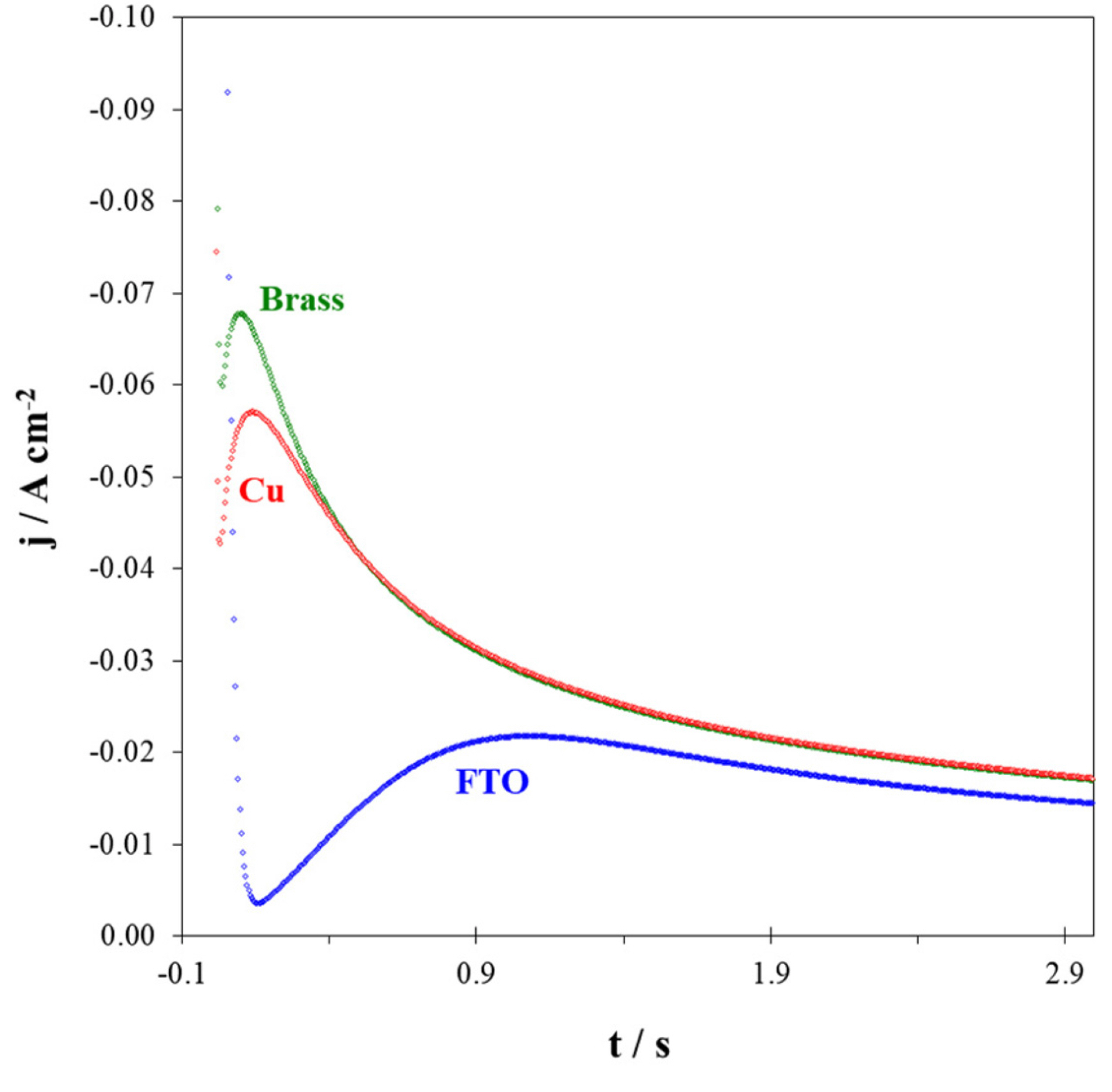
As a matter of knowledge, shape and size of the grains strongly depend on the nucleation and growth mechanisms [38,40,41]. Consequently, step-potential chronoamperometry technique is employed to evaluate the nucleation and growth mechanisms as well as the number of active nucleation sites. Fig. 5 shows the current transient curves corresponding to Fe-Pd electrodeposition at E = ŌłÆ0.95 V. General trend of these curves implies that after an abrupt drop in current related to double layer charging, the current increases due to the nucleation and growth. The current maximum observed at each transient results from the overlap of growing particles and/or their diffusion zones [1,40-44].
Fig.┬Ā5.
Current transient during electrodeposition of Fe-Pd alloy on different substrates; E = ŌłÆ950 mV vs. SCE.
In order to study the nucleation and growth mechanisms, different models can be applied. It should be pointed out that the well-known theories in the case of electrochemical nucleation and growth have been developed for single metal and/or single phase electrodeposition, such as Scharifker et al. [45,46] and Heerman-Tarallo [47,48] models. However, a literature survey shows that in many cases these models have been successfully utilized not only for binary alloy [1,6,16,49-61] but also for ternary alloy electrodeposition [62-64]. The well-known model of Scharifker and Hills [45] has been used for many multicomponent electrodeposition systems such as Fe-Ni [1,59], Fe-Co [50,60], Fe-Zn [61], Pd-Rh-Ru [62] and Fe-Pd [6]. In addition, our previous work shows that Scharifker and Hills model can well describe the electrochemical nucleation and growth of Fe-Pd [16].
In this model, it is assumed that nucleation is initiated at susceptible energetic sites such as dislocations, steps, kinks, grain boundaries, nubs, and etc [42,49,65,66]. According to AvramiŌĆÖs theory, the number of active nucleation sites (N) as a function of time (t) is estimated as:
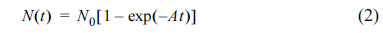
where A and N0 are the constant rate of nucleation and the number of final nuclei [65] (or number of active sites on the surface [49,66]), respectively; which depend on the electrolyte and cathodic potential [45,46]. When At is large enough (N(t) Ōēģ N0), nucleation occurs instantaneously and all nuclei are formed at the same time. As a result, there is a uniform distribution of the same size grains over the surface. Contrary, when At is too small (N(t) Ōēģ AN0t), the nucleation is progressive; i.e. new nuclei are formed whilst the previous created nuclei are growing. The progressive nucleation results in a lower number of nuclei and different grain size [65]. In order to identify the instantaneous and progressive nucleation, j-t transients (Fig. 5) are normalized in terms of maximum current (jmax) and its corresponding time (tmax). The mathematical relations of jmaxŌłÆtmax in the case of progressive and instantaneous nucleation are [45]:
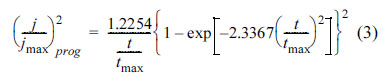
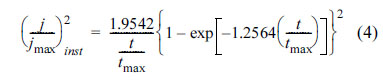
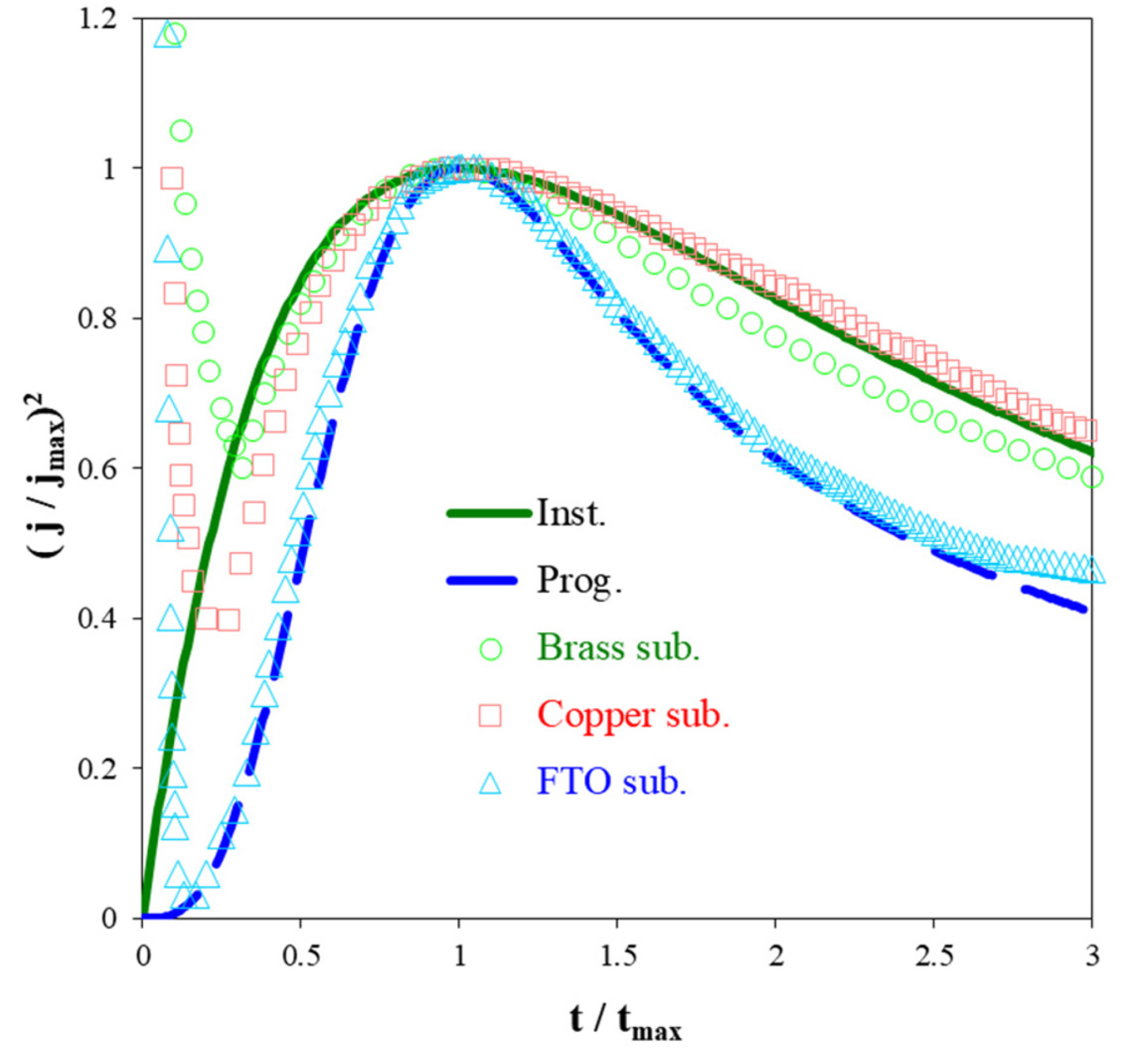
Fig. 6 shows the dimensionless forms of the experimental transients as well as standard curves corresponding to 3D nucleation and diffusion-controlled growth. As seen, in the case of brass and copper the nucleation process is instantaneous, while for FTO, it is progressive. Furthermore, the surface morphology shown in Fig. 4 is another evidence for validity of the utilized theoretical model.
Fig.┬Ā6.
Non-dimensional current transient ( j/jmax)2 vs. (t/tmax) during electrodeposition of Fe-Pd alloy on different substrates; E = ŌłÆ950 mV vs. SCE, solid line: instantaneous model, dashed line: progressive model, symbols: experimental results.
For instantaneous nucleation, N0 can be calculated using Eq. 5:
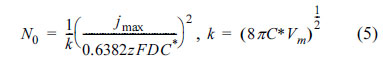
where C* and Vm are the bulk concentration of electroactive ions and the molar volume of deposit, respectively. Also, D is the diffusion coefficient in solution which is calculated as:

On the other hand, in the case of progressive nucleation, AN0 can be estimated as:
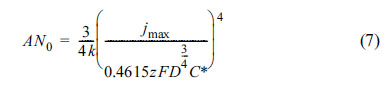
Here, D is obtained by

Finally, the number of saturated nucleation sites (Ns) can be estimated using:

In Table 2, the average grain size of the deposits (derived from Fig. 4) and the number of active nucleation sites (estimated from Eq. (5-9)) are presented. The minimum and maximum values of N0 correspond to FTO and brass, respectively. This result is in good agreement with the trend observed in SEM micrographs (Fig. 4); i.e. the deposit grain size on brass is much smaller (average 86 nm) compared to FTO (average 283 nm) due to its higher number of active nucleation sites. To sum up, estimation of the number of active nucleation sites is a first clue for the prediction of deposit grain size [40,41,50]. However, the final grain size also depends on the film growth rate [67,68].
3.3. Effect of overpotential on morphology and structure of alloy films
Figs. 7, 8 display the micrographs of electrodeposited Fe-Pd films on brass and FTO substrates at different potentials. It is observed that by an increase in cathodic potential from, the average grain size of the deposits increases (from 56 to 176 nm for brass and from 260 to 381 nm for FTO). Additionally, the same increase in potential results in a surface with higher roughness and porosity, and lower uniformity.
Employing chronoamperometric curves and regarding Eq. (5-9) for different potentials, the number of active nucleation sites (N0 in the case of brass and copper, and Ns in the case of FTO) are calculated for all substrates (Table 3). A comparison between the results presented in Figs. 7, 8 and Table 3, reveals that despite an increase in the number of active nucleation sites (due to increasing the overpotential), the grain size of the deposits becomes larger. As mentioned before, the final grain size of the deposits is influenced by both the number of active nucleation sites and the film growth rate. The important point is that higher overpotential will result in larger grain size if the rate of film growth prevails. Fig. 9 shows current transient curves corresponding to Fe-Pd electrodeposition on brass. With an increase in overpotential, the current maximum (jmax) shifts to more cathodic values and, accordingly, the number of active nucleation sites (Eq. 5) rises. However, this increase in overpotential leads to higher steady state current density (jss) as seen in Table 3. Considering this point, that jss is directly proportional to film growth rate, it is very likely that the film growth rate prevails on the high number of active nucleation sites. Therefore, despite higher number of nucleation sites due to increasing overpotential, the grain size of deposits becomes larger.
Fig.┬Ā7.
AFM micrographs of Fe-Pd electrodeposited films on brass; (a) E = ŌłÆ900, (b) E = ŌłÆ950, (c) E = ŌłÆ1000 and (d) E = ŌłÆ1050 mV vs. SCE.
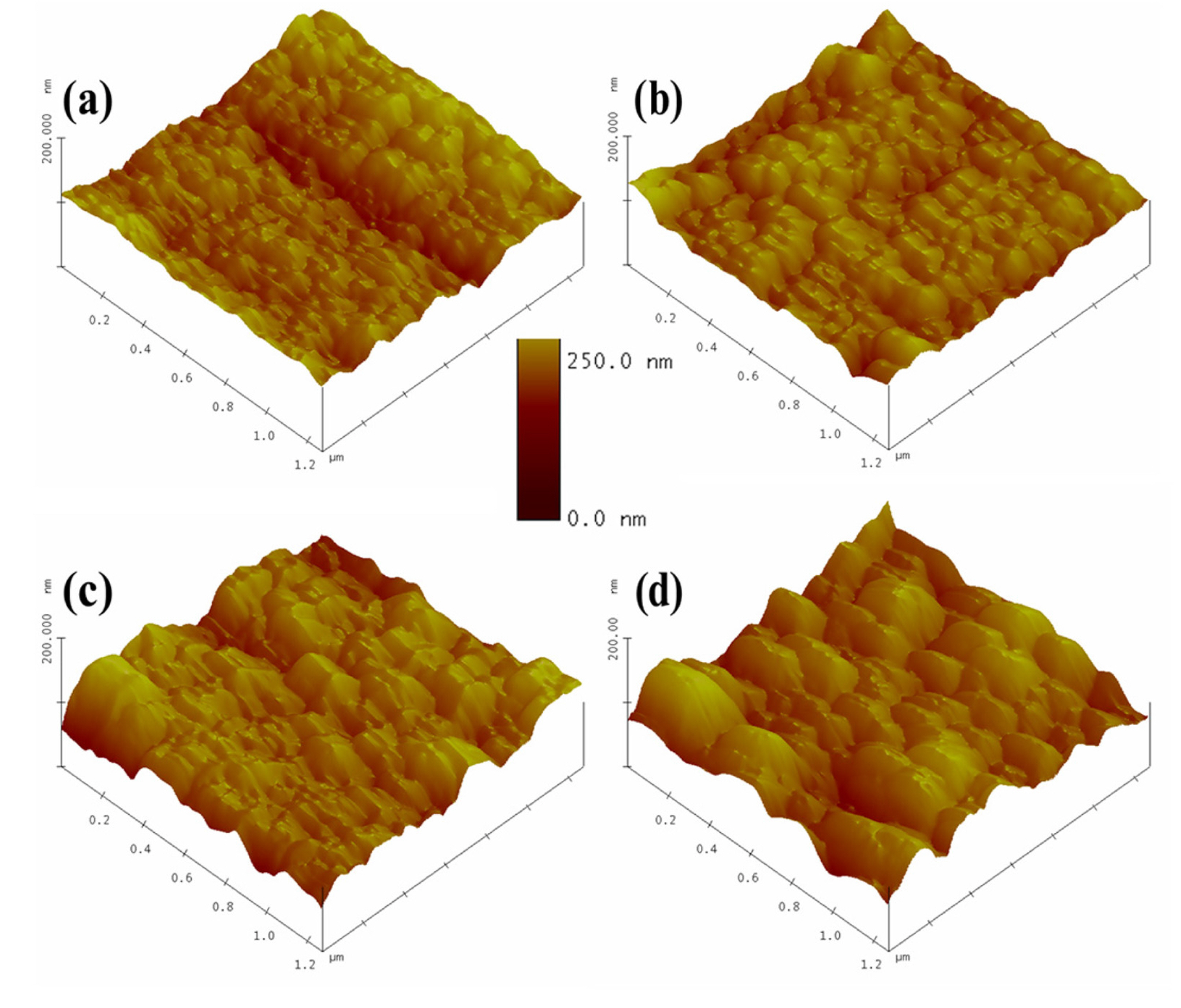
Fig.┬Ā8.
FESEM micrographs of Fe-Pd electrodeposited films on FTO. (a) E = ŌłÆ900, (b) E = ŌłÆ950, (c) E = ŌłÆ1000 and (d) E = ŌłÆ1050 mV vs. SCE. The scale bar represents 500 nm.

Table┬Ā3.
Effect of substrate and potential on the number of active nucleation sites and steady state current density.
Fig.┬Ā9.
Current transient during electrodeposition of Fe-Pd alloy on brass; E (from up to down) = ŌłÆ900, ŌłÆ950, ŌłÆ1000, ŌłÆ1050 vs. SCE.

X-ray diffraction studies were carried out to analyze various Fe-Pd deposited films at different potentials. According to the diffraction patterns shown in Fig. 10, in whole range of potentials, a sharp peak ( ) appears around 2╬Ė of 41.2┬░. This peak is almost similar to the standard diffraction peak of ╬│-Pd with face-centered cubic, but shifted to the high degree side, which can be attributed to the ╬│-Pd solid solution [20]. When the potential reaches ŌłÆ950 mV, a relatively weak peak (
) appears around 2╬Ė of 41.2┬░. This peak is almost similar to the standard diffraction peak of ╬│-Pd with face-centered cubic, but shifted to the high degree side, which can be attributed to the ╬│-Pd solid solution [20]. When the potential reaches ŌłÆ950 mV, a relatively weak peak ( ) 2╬Ė of 43.8┬░ appears as a second phase while ╬│-Pd phase lines gradually weaken. The peak around 2╬Ė of 43.8┬░ is almost similar to the standard diffraction peak of Fe or that of ╬▒-Fe with body-centered cubic structure, but shifted to the low degree side, which can be attributed to the ╬▒-Fe body centered-cubic solid solution [7,18].
) 2╬Ė of 43.8┬░ appears as a second phase while ╬│-Pd phase lines gradually weaken. The peak around 2╬Ė of 43.8┬░ is almost similar to the standard diffraction peak of Fe or that of ╬▒-Fe with body-centered cubic structure, but shifted to the low degree side, which can be attributed to the ╬▒-Fe body centered-cubic solid solution [7,18].
) appears around 2╬Ė of 41.2┬░. This peak is almost similar to the standard diffraction peak of ╬│-Pd with face-centered cubic, but shifted to the high degree side, which can be attributed to the ╬│-Pd solid solution [20]. When the potential reaches ŌłÆ950 mV, a relatively weak peak ( ) 2╬Ė of 43.8┬░ appears as a second phase while ╬│-Pd phase lines gradually weaken. The peak around 2╬Ė of 43.8┬░ is almost similar to the standard diffraction peak of Fe or that of ╬▒-Fe with body-centered cubic structure, but shifted to the low degree side, which can be attributed to the ╬▒-Fe body centered-cubic solid solution [7,18].Fig.┬Ā10.
X-ray diffraction patterns of electrodeposited Fe-Pd alloy on brass at different applied potential. S,  and
and  represent the substrate, ╬│(111) FCC and ╬▒(110) BCC phases, respectively.
represent the substrate, ╬│(111) FCC and ╬▒(110) BCC phases, respectively.
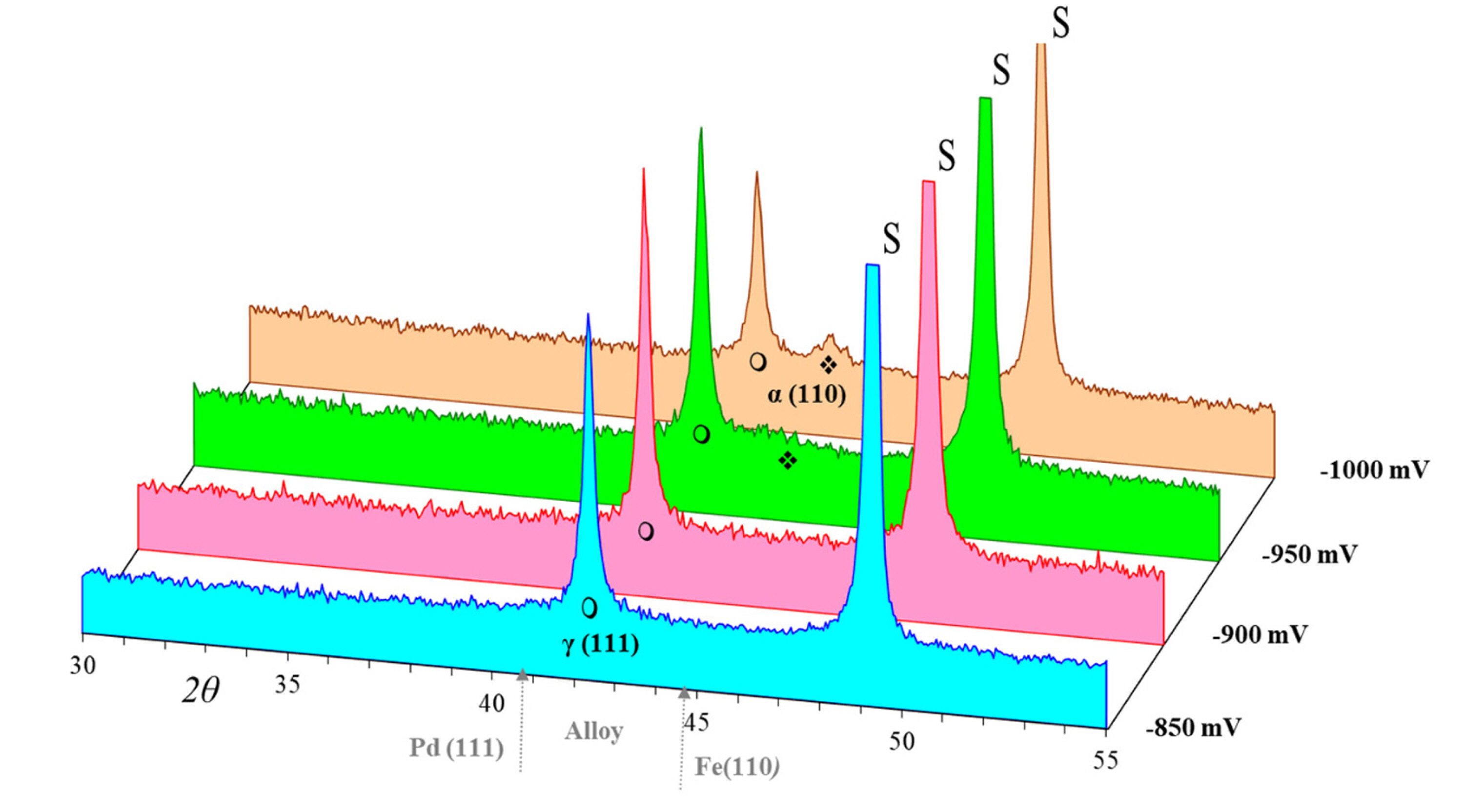
Using Debye-Scherrer formula [69], the crystallite size of the deposited films is estimated to be 31, 28, 22 and 19 nm for potentials of ŌłÆ850, ŌłÆ900, ŌłÆ950 and ŌłÆ1000 mV, showing nanostructured alloy films. These results reveal that by the potential shift to more cathodic values, the crystallite size of the deposits decreases. A comparison between the crystallite and grain size (56 to 176 nm) reveals that each grain consists of some crystallites [70], and contrary to grain size, crystallite size of the deposits decreases with an increase in overpotential. Therefore, it is likely that the crystallite size of the deposits is affected by the number of active nucleation sites while the grain size depends on both number of nucleation sites and film growth rate.
4. Conclusions
The voltammetric analysis showed that Fe-Pd electrodeposition is a diffusion-controlled process. Chemical composition of the deposited films implied a typical anomalous co-deposition on different substrates in which an increase in overpotential leads to higher Fe content. Cathodic current efficiency is low due to high catalytic activity of Pd toward H2 evolution. Morphological features of the thin films revealed that the grain size of the deposits is too small in the case of copper and brass in compared to FTO. Current transient curves indicated a 3D nucleation and diffusion-controlled growth. Using theoretical models, the number of active nucleation sites was calculated and considered as the reason for formation of nanostructured films. The grain size of the deposits becomes larger by increasing the overpotential because film growth rate prevails on the high number of nucleation sites.




