1. Introduction
Electrochemical energy storage devices were used to store the produced energy and discharge it at the time of use. Electrochemical secondary batteries; fuel cells and supercapacitor are the three main types of electrochemical storage devices each having its own advantages. Supercapacitors also have distinctive advantages such as high-power density, long cycle life, rapid charge and discharge, safe operation and power delivery at extreme climatic conditions [1ŌĆō3].
The key issues of supercapacitors are their low energy density. Till now commercially available supercapacitors can only provide an energy density of less than 10Wh/kg. Whereas, Lithium ion batteries can provide an energy density of more than 180 W h / kg. Therefore, research works are being carried out to enhance the energy density of supercapacitors by developing new electrode materials, novel electrolytes with a wide operation voltage window, or an ingenious device design. Several types of supercapacitors can be distinguished, depending upon the charge storage mechanism as well as the active material type. Electric double layer capacitors (EDLC) use activated carbon materials as a electrodes and stores the charge electrostatically by reversible adsorption of electrolyte ions onto active materials that are electrochemically stable having large specific surface area, on which the charge is separated on polarization at the electrodeŌĆōelectrolyte interface.
Pseudo-capacitors or redox supercapacitors use transition metal oxides as well as electrically conducting polymers and electric energy is stored by Faradic redox reactions between the electrolyte ions and the electrode, thus forming capacitance. The electrode materials play a vital role in the electro capacitive mechanism of the supercapacitors. The electrode materials are characterized based on their specific capacitance, the specific surface area, pore size and geometry and electronic and ionic conductivity. Carbon based materials tend to have better cycle stability and rate capability thus the supercapacitors can be operated at high charge and discharge rates with a lifetime of over a million cycles [4,5].
The value of specific capacitance of carbon materials strongly depends on its microstructure. The electric double layer formation and ion adsorption processes take part pores with diameter close to diameter of adsorbed ions, mainly micropores (d<2 nm). Whereas, the mesopores (2<d<5 nm)and macropores (d>5nm) facilitate the transport of the ions through the inner structure of the electrode. Recently, carbon materials including graphene carbon nanotubes and amorphous carbon have been widely investigated owing to their easy accessibility, non-toxicity and high chemical stability [6,7].
In general, these carbonaceous materials show a higher ion storage capacity than the theoretical capacity of graphite. However, these nanostructured carbons always have disadvantages of high production cost and complicated preparation processes and environmental concerns. This urges researchers to have more environment-concerned design, synthesis and characterization. For this natural biomass derived carbons emerges as excellent alternatives in substituting conventional carbon materials towards a wide range of applications [8].
The carbons produced from the biomass materials are naturally porous or hierarchical structured which can facilitate the electrolyte penetration and reduce the ion diffusion distance. Most natural biomass materials contain nitrogen, boron and other elements, which can be doped as heteroatoms, allowing generating of extra active sites [9,10]. Some bio-masses are recycled from the agricultural or daily wastes such as bamboo chopsticks [11],orange peel [12,13],wood saw dust [14,15],sugarcane bagasse [16], which makes the process cost-effective and environment friendly. Many biomass materials have been prepared from plants such as peanut shell [17], rice husk [18,19],corn stover [20,21], bamboo [22ŌĆō24], willow catkins [25],fishtail palms [26]. Since biomass in earth is abundant, renewable and low-cost biomass derived materials are being exploited for various applications, such as CO2 capture, hydrogen storage, water treatment, catalysis and energy storage. To convert the biomass into carbon, different methods such as carbonization are using air, steam, carbon dioxide and activation methods using physico- chemical methods. Ultrasonic assisted methods are recently emerging method to improve the quality of the materials prepared [27]. The objective of the work is to convert the biomass into carbon material for the electrochemical energy storage applications.
2. Materials and Methods
2.1 Biomass Materials
The waste corn cob is collected from the local market and grind to reduce the size. The powder form of the biomass is taken for further experiment. This is used as precursor to produce activated carbon.
2.2 Preparation of ultrasonic assisted chemically activated Carbon (UCA)
7.8 grams of raw corn cob (CC) powder (average particle size 0.47 mm) and 25 ml of concentrated H2SO4 (98%) was mixed together and kept for about 8 hrs. The dehydrated corncob powder was washed thoroughly with distilled water and 1M NaOH until the pH of the supernatants becomes 7. The material was dried at 85┬░C using hot air oven for about 6 h then ground and sieved. This material is labeled as chemically activated corncob (CA). About 2 g of activated corncob was taken in the 50 ml of distilled water. This mixture was continuously sonicated with an ultrasonic working frequency of 33┬▒3 kHz for 3hr using bath type sonicator and mechanically agitated with a speed of 550 rpm for 1 h. The treated corncob slurry was separated by filtration and dried at 90┬░C for 24 hrs and ground and the average particle is measured as 0.345 mm. This material is labeled as UCA and stored in a small glass container. The schematic diagram of the process is given in Fig. 1 (a).
2.3 Preparation of ultrasonic assisted physically activated carbon (UPA)
About 1 g of sieved corn cob powder (average particle size 0.46 mm) was taken in a ceramic boat with a lid, and they were then placed inside a muffle furnace. The temperature was maintained at 700┬░C for 8 min[28]. At last, the sample was cooled to room temperature without controlling the temperature. The pre-carbonization reaction of the biomass is given below [29].
After the high temperature pyrolysis of the corn cob 0.68 g of the sample along with 0.06 g of D-Tyrosine was dissolved in 50 ml of chloroform, and the mixture was sonicated for 30 minutes in an ultrasonic bath. High speed centrifugation (5000 rpm, 5 min) was applied to the mixture to precipitate amorphous carbons. After these procedures, the suspended carbon material is filtered and is dried in an oven. The carbon obtained is labeled as UPA. The schematic diagram of the process is given in Fig. 1(b).
2.4 Preparation of Manganese and nitrogen doped carbon (Mn/N-C)
About 0.3 g of UPA sample and few drops of ammonia (25%) is added, while stirring till pH raises to 10 and 5 ml of hydrazine hydrate is added for simultaneous reduction and nitrogen doping. Then it is washed with hydrochloric acid (5%) and water; dried and labeled as N-C. About 0.340 g of the nitrogen doped carbon material and 1.348 g of potassium permanganate is added and stirred for about 3hrs while the temperature is maintained at 100┬░C. The color of the solution turns to brown color indicates the decomposition of KMnO4 as below [30].
Then the solution was sonicated for about 3 hr. Then the solution is washed with ethanol and water and dried at 60┬░C and labeled as Mn/N-C.
2.5 Preparation of the working electrode
The glassy carbon (GC) disk electrode with the area of 0.037 cm2 was considered as the working electrode. Prior to use, the GC was polished with alumina powder till to get mirror finish. To prepare the working electrode, sample slurry was made by ultrasonically dispersing 150 mg of the sample in 150 ╬╝L N-methyl pyrrolidone, 100 ╬╝L ethanol and 0.1 ml isopropyl alcohol mixture with 5 wt% Nafion solution for 20 min. Then, 2.5 ╬╝l of the sample slurry was coated on to the top of the GC electrode and dried at room temperature to obtain mass loading of 0.0785 g/cm2.
3. Results and Discussion
3.1 X-ray diffraction analysis (XRD)
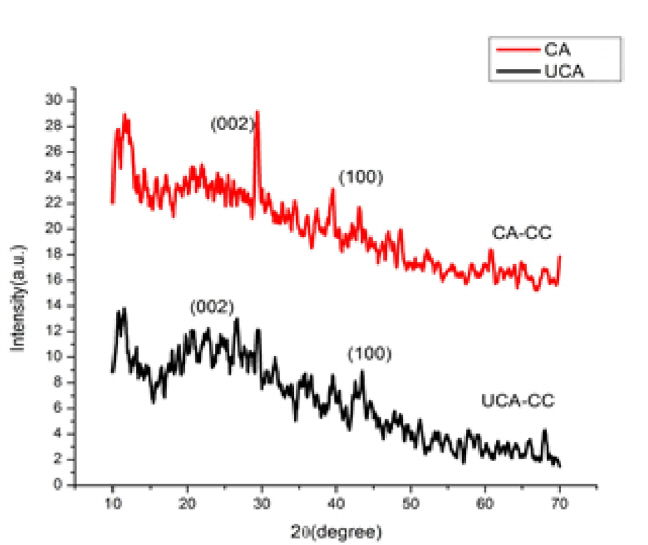
X-ray Diffraction patterns were carried out using (M/s GE Inspection Technology) by step scan technique with Cu-K╬▒ radiation (1.5405 ├ģ, 40 kV, 30 mA and scanning speed of 2┬░ in 20 minŌłÆ1) is shown in the Fig. 2. The peak positions were measured, and BraggŌĆÖs law was applied and the d spacing was calculated using eq. (1).
Where, n = integer 1, 2, 3... (Here n=1), ╬╗ = wavelength in angstroms (1.54 ├ģ for Cu), d = interatomic spacing or d spacing and ╬Ė = diffraction angle in degrees. The XRD patterns were analyzed for the structural parameters using the classical Debye Scherer formula [16].
Where, Lc is the crystallite height, interlayer spacing or stacking height (├ģ), La is the crystallite diameter, microcrystalline diameter or lateral size of the crystal, ╬╗ is the X-ray wavelength (nm),╬Ė is the Bragg diffraction angle (degree) and ╬▓ is the peak width at half maximum(radians(R) of ╬Ė, R = 0.01745╬Ė ┬░). But the calculated Lc and La values are not exactly equal to the stacking height and lateral size of the crystallites because these equations are derived for highly graphitized carbons and are not suitable for turbostratic carbons. Therefore, these can be used as convenient relative estimates of actual stacking height and lateral size of the crystallites. The actual crystallite sizes, therefore, are likely to be slightly greater than that of the calculated values. In the case of ultrasonic assisted chemical activated carbon diffraction within the range 2╬Ė = 22ŌĆō30┬░, from Fig. 2 sample UCA shows more developed and separated peaks with maxima at 2╬Ė =23, 26 and 29┬░ compared to chemical activated carbon and corresponding mean d002 spacing of 3.85, 3.39 and 3.07 ├ģ, respectively. The second diffraction profiles for both the samples were in the range around 2╬Ė = 43┬░, are diffuse and broad with weaker intensity. This indicates that the carbon layers are less developed. Some diffraction peaks are observed in the low range of 2╬Ė = 14ŌĆō22┬░, which gradually reduced upon ultrasonic treatment. These were attributed to residual cellulose crystallites this means that the attack of H2SO4 on the lignocelluloses does not lead to complete destruction in the primary stages, of the constituent cellulose crystallites and hence the sharp peaks are found in the range of 2╬Ė = 14ŌĆō22┬░. The unusual peak at 29┬░ may be due to pseudocrystalline structure formed due to the action of sulfuric acid. The calculation of the Lc and La values for chemically activated carbon and ultrasonic assisted chemically activated carbon at the angular values of 2╬Ė = 14ŌĆō30 and 43┬░, showed that the apparent crystallite heights (Lc) ranged between 40 and 70 ├ģ, and lateral dimensions (La) of 25ŌĆō40 ├ģ. It is noticed from the analysis both CA and UCA samples shows turbostatic natured carbon.
3.2 Fourier transforms infrared spectroscopy (FTIR)
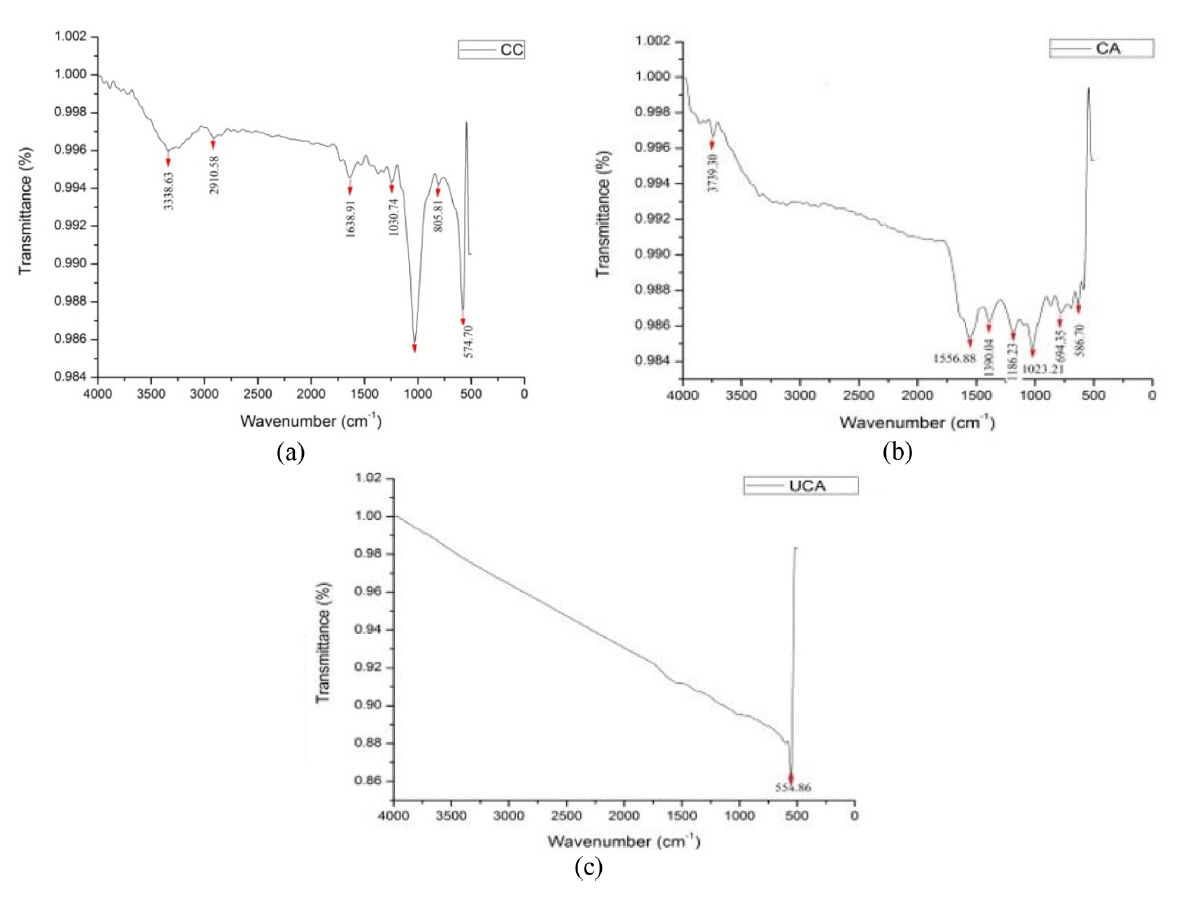
Functional groups of raw corncob (CC), chemically activated corncob (CA) and ultrasonic assisted chemically activated corncob (UCA) constituents were determined using FTIR spectroscopy model (M/s. Bruker vector 22 instrument) and is given in the Fig. 3a, 3b, 3c. The resulting spectrum represents the molecular absorption, creating a molecular fingerprint of the sample. Like a fingerprint no two unique molecular structures produce the same infrared spectrum. This makes infrared spectroscopy useful for several types of analysis [31]. The mid-infrared spectrum (4000ŌĆō400 cmŌłÆ1) is approximately divided into four regions. It is obvious from Fig. 3a that the peak at 3303 cmŌłÆ1 corresponds to O-H stretching vibrations that indicates the presence of hydroxyl groups; while that near 2844 cmŌłÆ1 depicts C-H stretching that corresponds to the presence of alkanes. 1000 cmŌłÆ1 depicts C-O stretching, with the peak near 600 cmŌłÆ1 showing characteristics of C-H bending. These functional groups represent the chemically active components of biomass. For the better understanding of the functional groups common to the structure of corn cob (CC). Table 1 presents the chemically active components related to the bonds of the atoms that make up the functional groups of the material which take part during chemical conversion processes [28,32]. During acid treatment the chemical decomposition of the cellulose content occurs due to ŌłÆOH groups which facilitates decarboxylation reactions (Fig. 3b), while the presence of C-H groups due to alkanes causes hemicellulose degradation [33]. The C=C bonding, due to the presence of alkenes, facilitates the lignin decomposition reactions; C-O bonds assigned to carboxylic groups in cellulose and hemicellulose, leads to the breakage of glycosidic bonds that consequently forms a series of less oxygen-containing compounds such as ethers, acids, and aldehydes and non-condensable gases such as CO and CO2 [2,14]. From Fig. 3c it is clear that the high energy ultrasonic waves through the acid treated biomass causes complete disruption in the chemical bonds and only a single peak is found at 554.86 cmŌłÆ1 due to unbroken C-H bonding. The ultrasonic assisted carbon material showed good characteristics.
3.3 Scanning Electron Microscopy
The SEM analysis is done to study the surface morphology of the sample materials using model (M/s. Carl Zeiss MA15/EVO 18 Scanning Electron Microscope). The results are given in Fig. 4 (a),(b) and (c). Where Fig. 4a, represents the SEM images of ultrasonic assisted chemically activated corn cob which shows distinct microporous nature of the sample material with pore size ranging from 4.68 to 2.68 ╬╝m. Microporous carbon electrode improves energy density by increasing capacitance (i.e., more adsorbed electrolyte ions during polarization) since micropores facilitates charge retention. Power capability is not improved because of slow dynamics of the pre-desolvation process and diffusion of electrolyte ions into the micropores. Fig. 4b represents the SEM images of ultrasonic assisted physically activated corncob which shows flaky and microporous nature of the sample material pore size ranging from ~4.91 to 2.5 ╬╝m. Fig. 4c represents the surface characteristics of Mn/N-C which has metal oxide deposits aggregated on the carbon sample and has reduced pore size.
3.4 Energy Dispersive X-Ray Spectroscopy
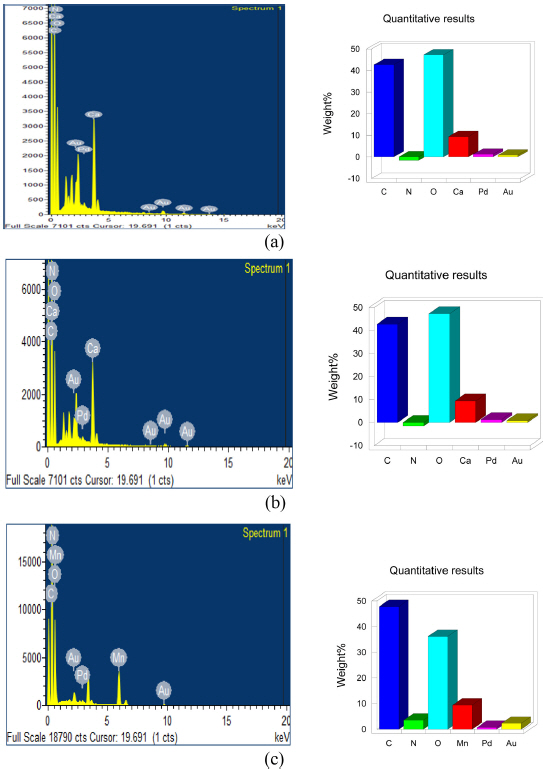
The EDAX analysis is an analytical technique used for the elemental analysis or chemical characterization of a sample. The analysis is done using(M/s. Oxford instruments nanoanalysis INCA energy 250 microanalysis). The results are given in Fig. 5a, 5b, 5c the presence of Au, Pd is due to vaccum sintering of the sample before the analysis.The analysis was done to find the various heteroatoms in the sample and the effect of pretreatment and ultrasonication on it. Fig. 5a, 5b shows the EDAX images of UCA,UPA respectively both the samples showed almost same elemental composition and the oxygen content in both the samples were 47.34 wt%. The nitrogen doping improves the electronic and chemical properties of the prepared carbon materials due to its atomic size and five valence electrons. It is noticed from the Fig. 5c that 3.53 wt% of nitrogen and 9.44 wt% of manganese have been doped to the sample and the oxygen content had dropped to 36.20 wt% due to nitrogen doping. This implies that nitrogen doping facilitates the corrosion resistance of material by reducing the oxygen containing groups.
4. Electrochemical Studies
The electrochemical studies were measured by (CHI660D) electrochemical workstation in a conventional three electrode system where reference electrode is Ag/AgCl electrode (E = +0.197V),working electrode is glassy carbon electrode, counter electrode is platinum wire and 1M sodium sulfate is used as electrolyte and the potential window is 0ŌĆō1 V.
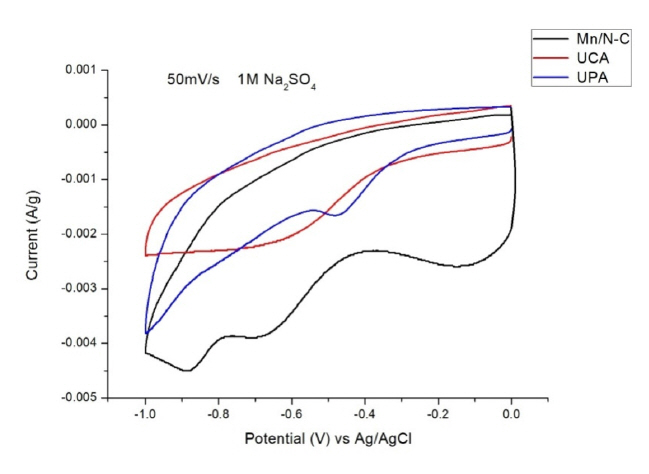
4.1 Cyclic Voltammetry
For cyclic voltammetry (CV) the value of specific capacitance (Csp) is calculated from eq.(7) [34,35]
Where, Q is the area under the CV curve, ΔV is the potential window (V),
╬┤ V ╬┤ t
Where, C+ denotes the cations.
4.2 Galvanostatic Charge/Discharge Studies
The galvanostatic charge/discharge (GCD) studies were performed at different current densities (taking current values from cyclic voltammetric curves) to analyze the capacitive behavior of prepared carbon materials. The voltammetric profile is very similar to those found in the literatures [36]. These processes are composed of several steps and the corresponding voltammetric peaks are overlapped and difficult to identify or to associate with a specific process. The charging current of the samples is symmetric to its discharge current, indicating the EDLC behavior during the charging and discharging steps. The specific capacitance (Csp) of the electrode material is calculated from the equation
Where, I is the constant discharge current (A), Δt is the discharge time (s), ΔV the potential change during the discharge process (V) and m is the mass of the active material on the electrode (g). The specific energy density (E) and power density (P) are calculated from galvanostatic charge and discharge graphs using eq. (9) &, (10) [37].
Where, Csp is the specific capacitance (F/g) and ΔV the potential change during the discharge process (V)
Where, E is the specific energy density (Wh/kg) and Δt the discharge time (s)
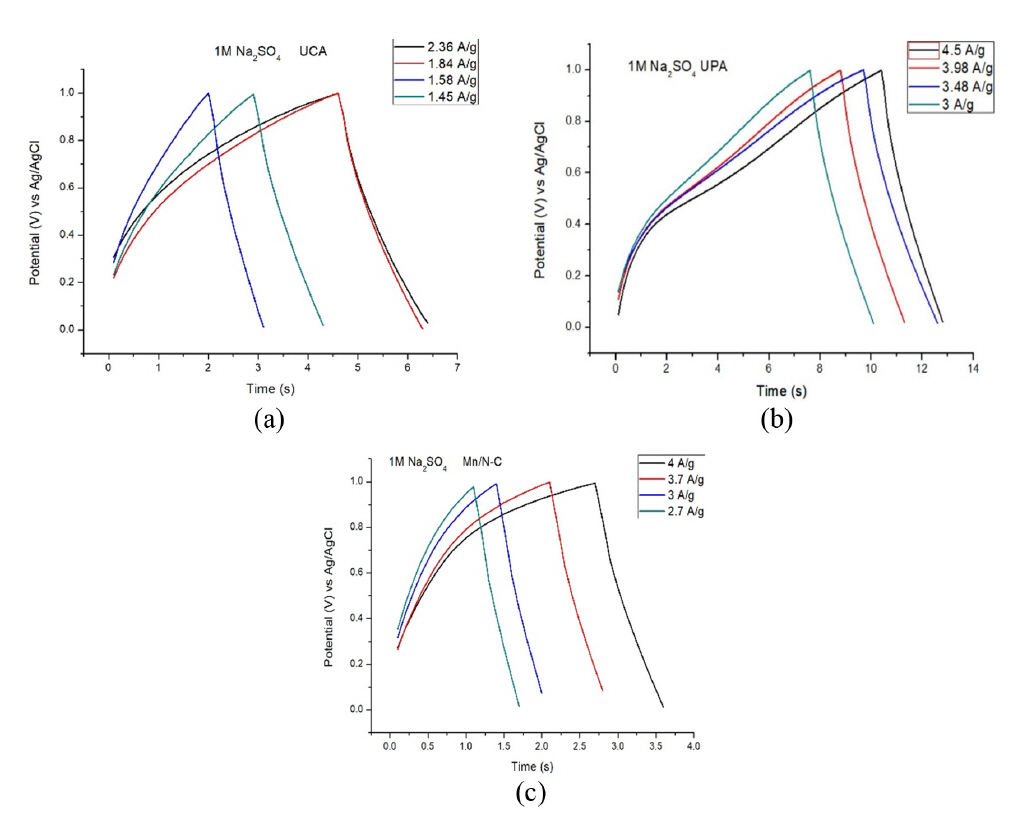
The capacitive performance of the samples was further tested with galvanostatic charge-discharge (GCD) experiments at various current densities with the voltage windows the same as that of the CV analysis. The GCD analyses of the three samples were given in Fig. 7a, 7b, 7c. The values of specific capacitance for the three samples at various current densities were given in Table 3, 4, 5. It can be observed from the table values that as the current density values of the sample increases the specific capacitance value decreases because at higher current densities there will be fast movement of ions and there wonŌĆÖt be enough time for the ion adsorption in the pores. From the galvanostatic charge and discharge (GCD) tests the values of specific capacitance values for UCA, UPA and Mn/N-C were found to be 162, 165 and 625 mF/g respectively at a current density of 0.175 A/g. The specific energy density values for UCA, UPA and Mn/N-C were found to be 2.40, 4.12 and 7.62 (Wh/kg) respectively. The power density values for the samples UCA, UPA and Mn/N-C were found to be 46.04, 87.97 and 131.42 W/kg respectively.
4.3 Electrochemical Impedance Studies
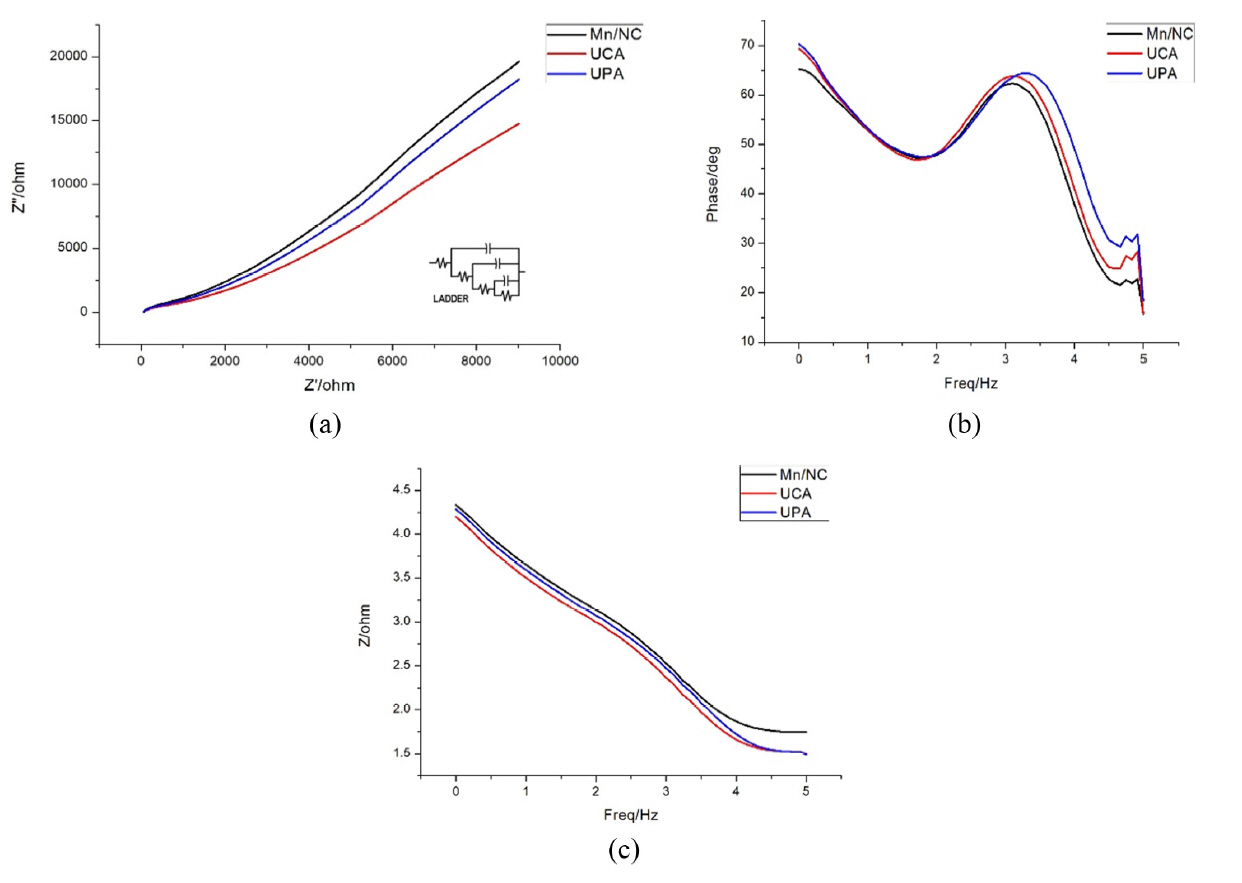
Electrochemical impedance studies (EIS) were conducted to examine the resistance characteristics of the carbon materials existing at the electrodeŌĆōelectrolyte interface. The experiment is done in frequency between 1 to 100000Hz under open circuit potential. The comparisons of the resistance characteristics for the three samples are given in Fig. 8a, 8b, 8c. The carbon materials show good capacitive behavior with nearly vertical slope at the low-frequency region. At very high frequency, the imaginary part (ZMM) of the impedance is close to zero, while the real part of resistance (ZM) such as the intercept of plot with real axis, represents the equivalent series resistance (ESR) which is a combination of the ionic resistance of the electrolyte, the intrinsic resistance of the activate materials and contact resistance with the current collector. The relationship between the total impedance and the frequency for the porous ACs are shown on Fig. 8c. When the frequency is low, the electrolyte ions try to penetrate from the orifice to the active material pores. So, the bottom of the pores will also devote to the resistive and the capacitive behavior that lead to higher impedance. However, at high frequency the electrolyte ions can only penetrate near the orifice of the pores. Hence, resistive and the capacitive behavior can only response near the orifice. The penetration degree decreases with the frequency increasing. At the frequency below 3.2 Hz, a straight line is obtained that is due to the accumulation of electrolyte ions at the bottom of pores. A horizontal line in the high frequency region signifies the dominance of electronic transport over this regime. The phase versus frequency plot (Fig. 8b) for the samples shows that at higher frequency the value phase value decreases. The EIS data are generally analyzed in terms of an equivalent circuit model (inset of Fig. 8a), whose impedance matches the measured data. However, the circuit components in this type of model are not assigned to the physical processes of the cell. The model chosen gives the best possible match between the model's impedance and the measured impedance.
5. Conclusions
The activated carbon materials were prepared from waste biomass by ultrasonic assisted chemical activation method, ultrasonic assisted physical activation method, and manganese nitrogen doped carbon. The cyclic voltammetry result shows that the specific capacitances were found to be 93, 100 and 115 F/g for UCA, UPA and Mn/N-C respectively. From the galvanostatic charge and discharge tests the values of specific capacitance were found to be 162, 165 and 625 mF/g at current density 0.175 A/g. The specific energy density values for UCA, UPA and Mn/N-C were found to be 2.40, 4.12 and 7.62 (Wh/kg) respectively. The power density values for the samples UCA, UPA and Mn/N-C were found to be 46.04, 87.97 and 131.42 W/kg respectively. The electrochemical impedance spectroscopy was done at low frequency between 1 to 10 kHz. The Nyquist plot gives the resistant characteristics of the materials due to diffusional resistance at the electrodeŌĆōelectrolyte interface. Among the three materials prepared manganese and nitrogen doped carbon shows good physical and electrochemical properties.