Introduction
Direct formic acid fuel cells (DFAFCs) are considered promising alternative power sources for portable power devices because of their significant advantages, including high theoretical open circuit voltage (1.45 V), low crossover through the polymer membrane, high fuel concentration, low toxicity, and ambient temperature operability [1,2]. A wide range of sources of formic acid production have been mainly from fossil feedstock, but recently, biomass and CO2 hydrogenat ion have been studied extensively [3].
The electrocatalysts for both the anode and cathode electrodes are the most important factors affecting the final performance of DFAFCs, and Pt- and Pd-based catalysts have been studied widely because of their excellent catalytic activity and long term stability. In particular, Pt is considered the most suitable electrocatalyst among all metals because of its excellent stability [4,5]. On the other hand, CO-poisoning on the Pt surface by the dehydration pathway of HCOOH oxidation degrades its catalytic activity, which limits its use. The strong CO adsorption energy at low potential is responsible for the abundant CO adsorbed on the Pt surface [6,7]. The introduction of second metals, such as Bi [8], Ir [9], and Ru [10], can enhance the tolerance of Pt from CO poisoning by promoting CO2 formation directly or facilitating the CO oxidation reaction, which is due to either the synergistic catalytic effect or electronic structural effect.
Bismuth (Bi) is considered the most efficient modifier of the Pt catalyst for the HCOOH oxidation because of its third-body effect and electronic effect on the Pt surface [11–14]. Herrero et al. reported that Bi-modified Pt exhibited improved electrocatalytic activity and stability because of the synergistic effects of Bi in cleaving the O-H bond and Pt causing C-H bond scission [14]. In addition, Bi can promote the direct pathway (dehydrogenation) of HCOOH, which can reduce the extent of CO poisoning of Pt. On the other hand, Bi can be easily leached out from the electrode surface or oxidized to Bi2O3 at potentials over 0.8 V (vs. Ag/AgCl) in acid solution, which means that the Bi-modified Pt catalyst is so unstable when used at high potentials [12,15]. Recently, Pt-based tri-metal electrocatalysts, including PtPdAu [16], PtCuBi [17], and PtRuSn [18], have been studied widely to overcome such problems. Pd can be a good choice for the third element of the PtBi catalyst because Pd is very stable under acid conditions because of its stable electronic structure. In addition, BiPd [19,20], PdPt [21,22], and BiPt [13] catalysts could prevent the poisoning of Pt by CO due to the less CO chemisorption by the downshift of the d-band center.
In this study, PtBi/C and PtBiPd/C electrocatalysts were synthesized by the irreversible adsorption of Pd and Bi on a 10 wt. % commercial Pt/C catalyst followed by the NaBH4 reduction. The irreversible adsorption is a simple preparation process that can avoid the complicated alloying procedures. In addition, alloy structures can be easily prepared without altering the bulk properties [10,11]. The introduction of Bi enhanced the direct formic acid oxidation over the indirect oxidation pathway. In addition, Pd increased the oxidation potential of Bi from 0.9 to 1.2 V (vs. Ag/AgCl), which resulted in highly improved electrocatalytic activity in HCOOH oxidation.
2. Experimental
2.1. Catalyst synthesis
The PtBi/C and PtBiPd/C electrocatalysts were synthesized by the irreversible adsorption of Pd and Bi ions to commercial Pt/C followed by the reduction of metal ions. First, a 10 mM Bi solution was prepared by dissolving bismuth oxide (Sigma-Aldrich, USA) with 20 ml of 0.1 M HClO4. To synthesize the PtBiPd/C, mixed solutions of 10 mM of Bi and 0.5 mM of Pd ions were prepared by dissolving bismuth oxide and Pd(NO3)2 (Sigma-Aldrich, USA) with 20 mL of 0.1 M HClO4. The above solutions were kept at room temperature for 10 days to stabilize them. The irreversible adsorption of Bi ions or Pd and Bi ions was performed by adding 50 mg of commercial 10 wt. % Pt/C (Alfar Aesar, UK) to the above solutions with stirring (400 rpm) using a hotplate stirrer for 10 hours at 40°C and then were reduced by 0.019 M NaBH4 (Dae-Jung Chemicals & Metals Co., Ltd, Korea) for 30 min. It was washed several times with 0.1 M HClO4 solution followed by a drying at 70°C for 3 h in a vacuum oven.
2.2. Instrumental analysis
X-ray diffraction (XRD PANalytical X’Pert Pro MPD, Malvern Panalytical, USA) of the commercial Pt/C, PtBi/C, and PtBiPd/C was performed using 40 kV Cu Kα radiation. The scanning rate and 2θ range was 10°/min and 0–100°, respectively. The morphology was examined by high-resolution transmission electron microscopy (HR-TEM, JEOL JEM-2100F, USA). X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi Thermo Fisher Scientific, USA) was performed using an Al Kα X-ray source. Cyclic voltammetry (CV) was recorded on a SP-50 (Bio-Logic, USA).
2.3. Electrochemical evaluation
Catalyst ink was prepared by mixing 0.5 mg of catalyst powder with 1 ml of ethanol and 1 mg of Nafion (5 wt. %, Sigma-Aldrich, USA). After 30 min sonication, 10 μL of the paste was spread on the GC electrode with a micropipette and dried using a hot air drier for 15 min.
Electrochemical tests were conducted at room temperature in a three-electrode-system using an Ag/ AgCl (sat. KCl) reference electrode and a Pt wire counter electrode. H2SO4 (0.5 M) was used as an electrolyte solution and the electrode was subjected to potential cycling from −0.2 V to 1.2 V with a 10 mVs−1 scan rate. The 0.1 M formic acid oxidation was performed in 0.5 M H2SO4 with potential cycling from −0.25 V to 1.2 V at 20 mVs−1 scan rate.
3. Results and Discussion
As shown in Fig. 1 and summarized in Table 1, the metal nanoparticles were anchored evenly over the carbon support and the particle size Pt/C did not change significantly after the irreversible adsorption of Bi, or Pd and Bi, which suggests that Pd and Bi are only slightly adsorbed on the Pt surface.
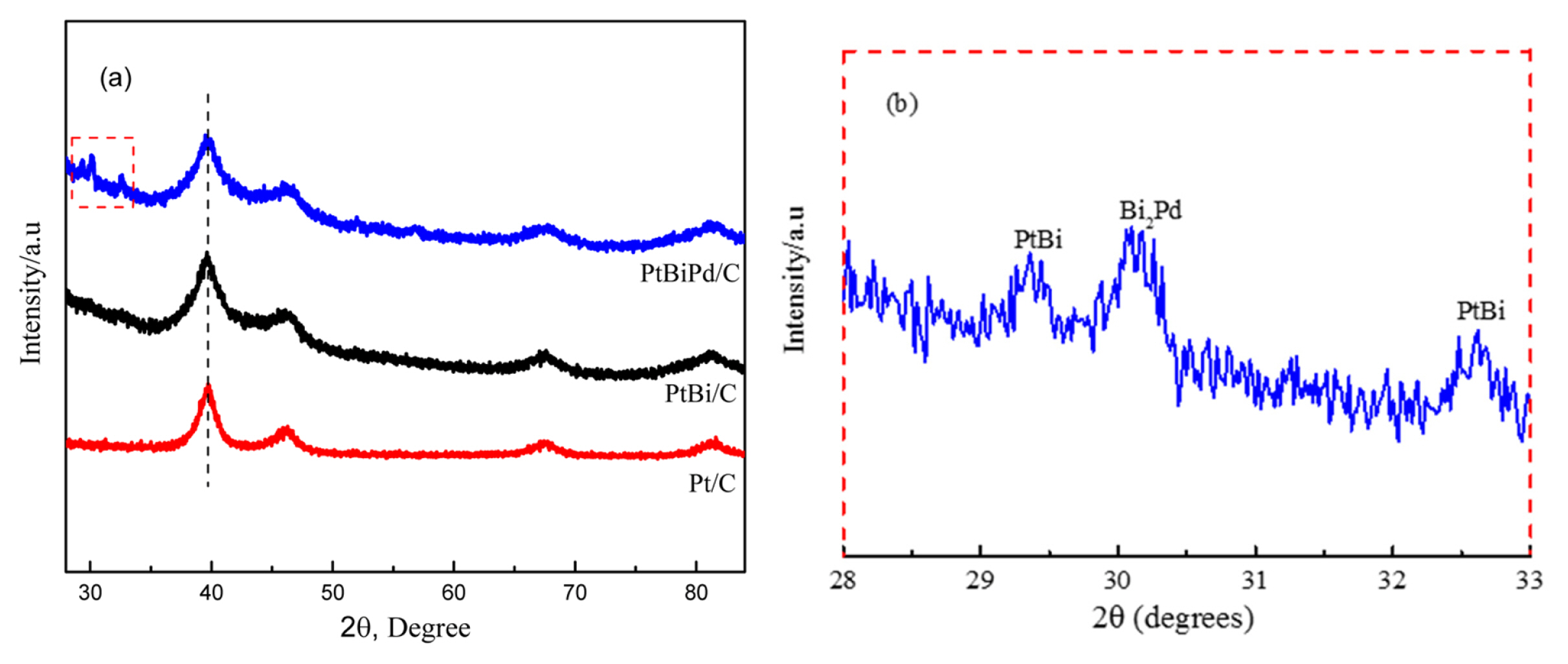
Fig. 2 presents XRD patterns of the Pt/C, PtBi/C, and PtBiPd/C. The broad XRD peaks at approximately 2θ = 40, 46, 67, and 80° were assigned to the Pt(111), (200), (220), and (311) planes, respectively. The XRD patterns of PtBi/C were similar because the characteristic peaks of Bi almost overlapped with those of Pt. On the other hand, the peak broadening suggests that a PtBi alloy is formed and partial Bi atoms incorporate into the Pt crystal lattice. In addition, when the Pd and Bi are co-adsorbed on the Pt/C, three small peaks are observed, corresponding to BiPt, and the second peak is consistent with Bi2Pd (Fig. 2b) [20,23]. The lattice parameters of the catalysts were calculated based on Bragg’s law and the Scherrer formula using the (111) XRD peak, as summarized in Table 2.
The lattice constant of the PtBi/C was larger than that of Pt/C because of the larger atomic size of Bi than Pt. Similarly, the smaller lattice constant of PdBiPt/C than that of PtBi/C can be due to the smaller atomic size of Pd than Pt and Bi. The variations of the lattice constant and crystallite size of various catalysts suggest the formation of alloy structures in the PtBi/C and PdBiPt/C catalysts [24].
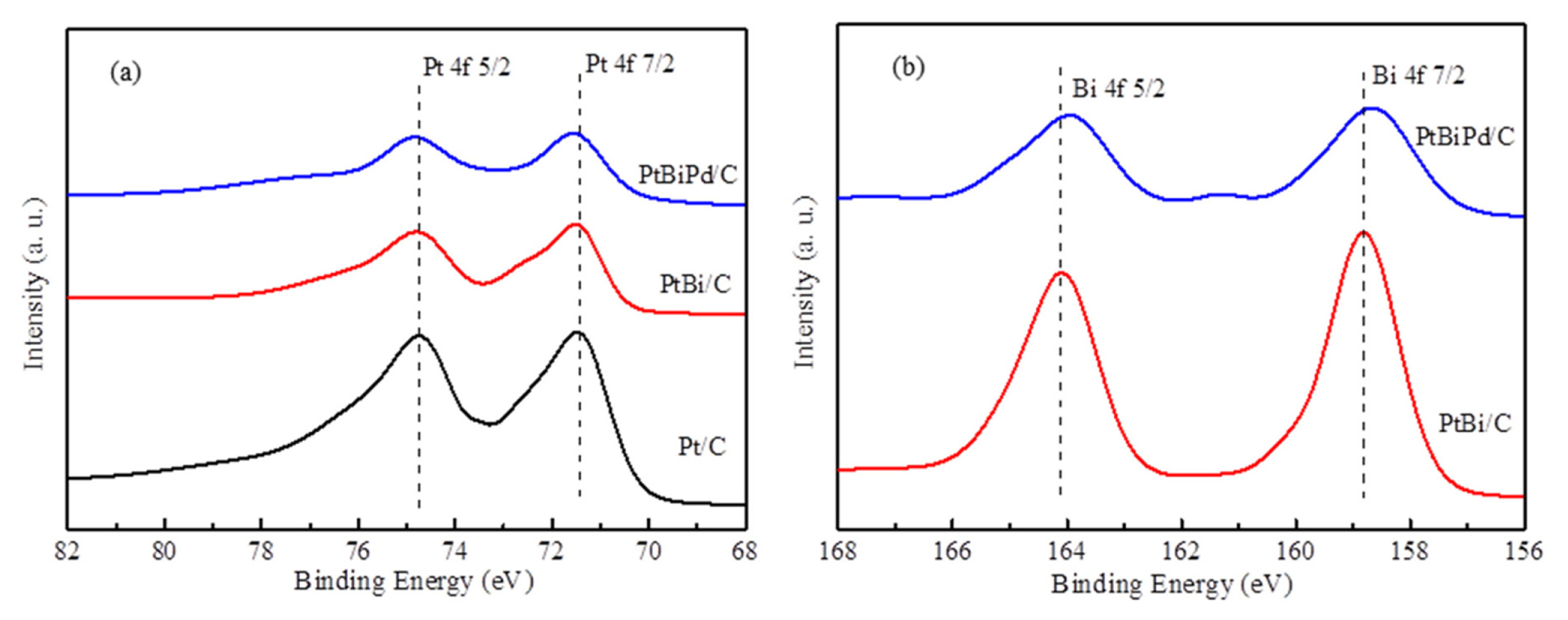
XPS was performed to examine the chemical structures of various catalysts. As shown in the XPS survey spectra of Fig. 3, Pt, Bi, and C peaks except for Pd were observed in each corresponding catalyst. Generally, it is very difficult to observe the Pd peak, particularly when the content is very small because its position overlaps with those of Pt and C. As shown in Figs. 4b–4d, the Pt 4f7/2 peak can be deconvoluted to two sub-peaks at approximately 71.5 and 72.9 eV, corresponding to Pt (0) and Pt(II), respectively [25]. The Bi 4f7/2 also can be deconvoluted into two sub-peaks at approximately 158 and 160 eV, which correspond to Bi(0) and Bi(III), respectively [26]. As summarized in Table 3, Bi(0) is dominant in both PtBi/C and PtBiPd/C, which indicates that Bi2O3 was almost reduced to Bi. Therefore, metallic Bi can be dominant on the catalyst surface. Interestingly, the Pt 4f peak shifted slightly toward a higher binding energy, as shown in Fig. 4(a), indicating the formation of an alloy structure between Pt and Bi. Similarly, the peak shift of Bi 4f to a lower binding energy indicated the formation of an alloy structure between Pd and Bi, as shown in Fig. 4(b) [27].
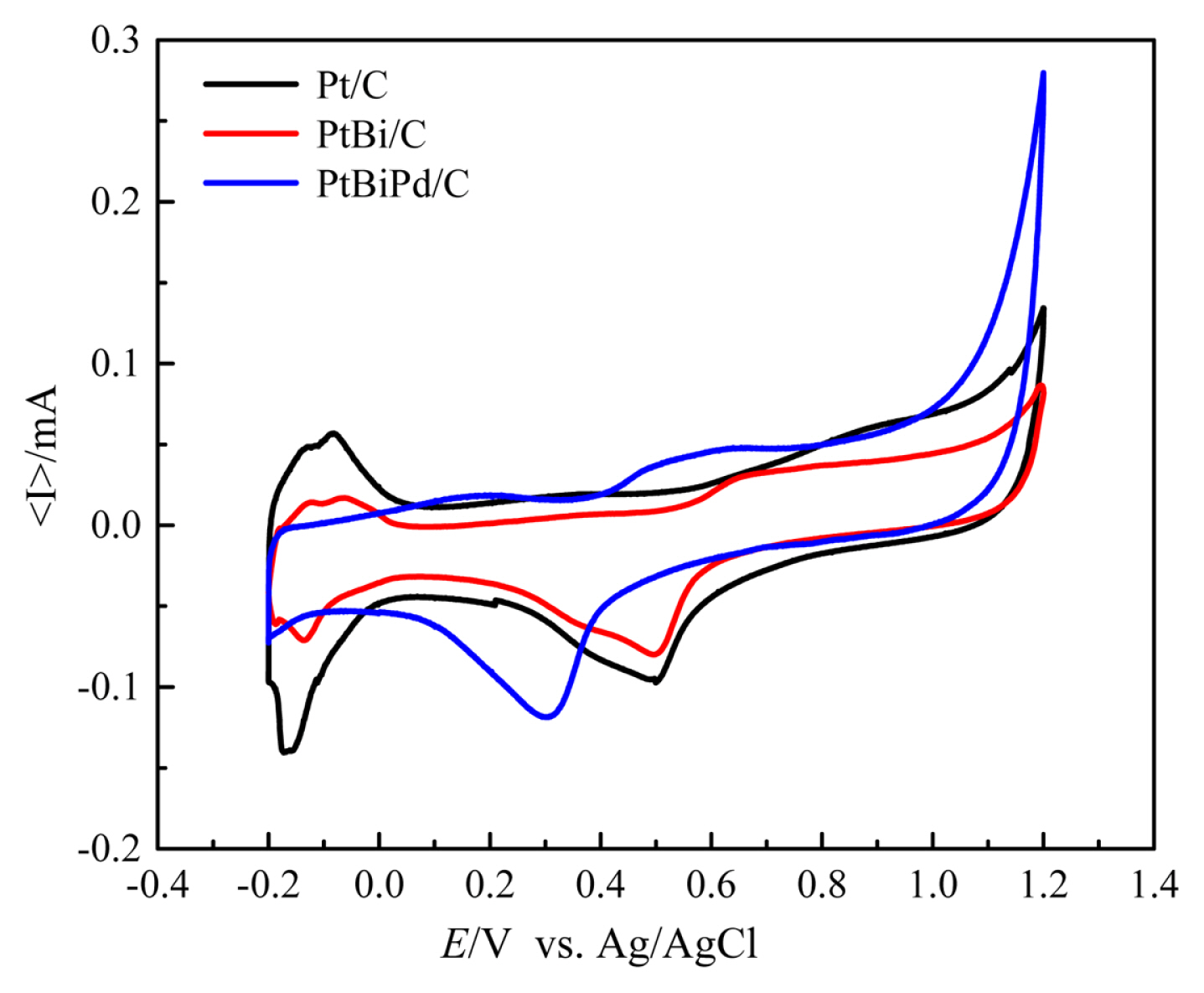
The electrochemical performance was examined by CV. As shown in Fig. 5, the hydrogen region below 0.0 V and Pt oxidation/reduction peak at ~ 0.8 and ~0.5 V were observed clearly for the Pt/C catalyst. When Bi was adsorbed on the Pt/C catalyst, the current related to the hydrogen region and Pt oxidation/reduction decreased because Bi atoms cover the Pt sites. The PtBiPd/C catalyst showed a further decrease in the current of the hydrogen region and a shift of the reduction potential to a lower potential than that of BiPt/C, which indicates alloy formation in the PdBiPt/C.
The formic acid oxidation reaction was performed using a range of catalysts in a 2 M HCOOH and 0.5 M H2SO4 solution. Generally, formic acid is oxidized to CO2 via two pathways [1].
As shown in Fig. 6, two oxidation peaks in the positive scan were observed, which correspond to the direct and indirect oxidation reaction pathways. The commercial Pt/C catalyst showed poor formic acid oxidation activity due to CO poisoning by the formation of COads on the Pt surface. Instead, the PtBi/C and PtBiPd/C catalysts exhibited enhanced electrocatalytic activity. As summarized in Tables 4 and 5, the current of direct formic acid oxidation (Id) increased ~ 8 and 16 folds for the PtBi/C and PtBiPd/ C catalysts compared to that of commercial Pt/C because of the electronic, geometric, and third body effects [20,27], which are higher values than those of previous studies [16,17]. In addition, the higher ratio between Id and the current of indirect formic acid oxidation (Iind) for the PtBi/C and PtBiPd/C catalysts indicates that the dehydrogenation pathway is dominant with less CO formation on these catalysts.
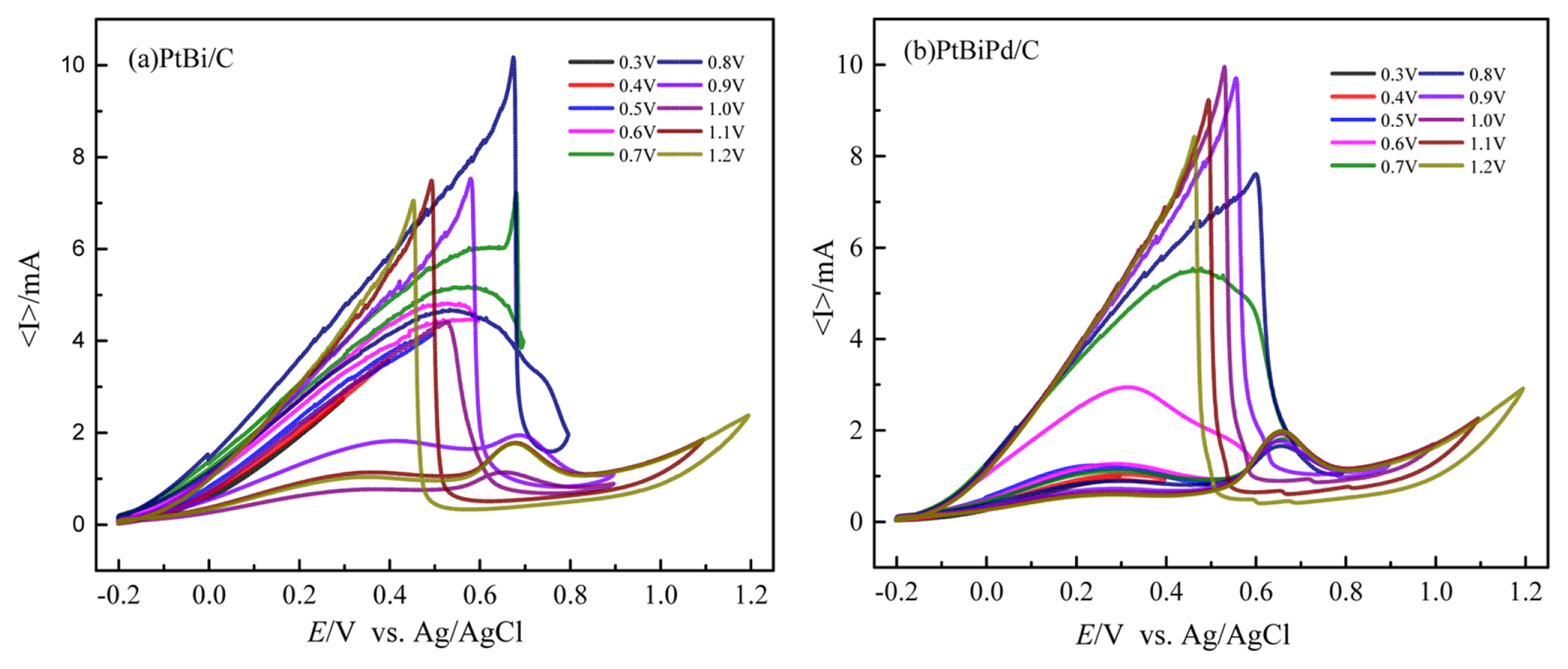
To examine the effects of Pd on formic acid oxidation, the scan range was varied, especially for the upper potential limit (UPL), because it can affect the reverse scan peak by the leaching of metal at high potential. As shown in Fig. 7(a), in the case of the PtBi/C catalyst, the maximum current of reverse scan increases initially with increasing UPL because of the oxidation of the COads at high potential. The reverse scan current was highest when the UPL was 0.8 V and decreased from 0.9 V, which is due to Bi leaching out at high potential [21,23,24]. Instead, in the case of the PtBiPd/C catalyst, the maximum reverse scan current was highest at 1.1 V of the UPL, which means that Bi leaching occurs at a higher potential than that of the PtBi/C catalyst by the co-adsorbed Pd [29], which can be attributed to the formation of the PdBi alloy structure that has a strong resistance against Bi leaching [30].
4. Conclusions
PtBi/C and PtBiPd/C catalysts were synthesized by the irreversible adsorption of Pd and Bi using Bi2O3, Pd(NO3)2 as precursors on commercial Pt/C followed by NaBH4 reduction. Instrumental analysis showed that the alloy structure was formed among Pt, Bi, and Pd. The Bi atoms enhanced the direct formic acid oxidation reaction and Pd atoms improved the resistance against Bi bleaching at high potential, which strongly improved the overall formic oxidation reaction.