1. Introduction
Currently, conventional lithium-ion batteries have gained immense attention as an energy source for various applications, from small devices to large-scale energy storage systems, including the electric transport sector. Lithium-ion batteries have outperformed all the other commercial battery technologies existing today owing to their high gravimetric and volumetric energy densities, high power rate capability, and long lifetime [1,2]. However, to further increase the energy density of lithium-ion batteries, the development of novel electrodes with high specific capacity is imperative. Graphite, which exhibits a moderate specific capacity of 372 mAh g−1, is a commonly used anode material [3,4].
Among the several electrode materials investigated for battery applications, phosphate-based electrode materials have gained considerable attention owing to their robust anions, which improve the safety, specific capacity, energy density, and rate capability of battery systems [5-9]. VOPO4-based materials with various structures including crystalline waters have been studied as cathodes for lithium-, magnesium-, and sodium-ion batteries [10-14]. In addition, VOPO4-based cathode materials with various structures can react reversibly with Mg and Li ions. Moreover, their reaction mechanisms for Mg and Li are different. The low gravimetric capacity of Mg (2205mAh g−1) as compared to that of Li (3861mAh g−1) can limit the durability and practical applications of magnesium battery systems [15]. Furthermore, vanadium phosphates have rarely been investigated as anode materials for lithium-ion batteries. Zhang et al. [16] prepared amorphous VOPO4 using the hydrothermal method with heat treatment for application as an anode material. It was found that the anode maintained a specific capacity of 300 mAh g−1. Ren et al. prepared LiVOPO4 with the β phase via a sol-gel method followed by heat treatment. The resulting anode showed a specific capacity of 380 mAh g−1 only for 30 cycles [17].
In this study, we investigated the electrochemical performance of VOPO4·2H2O with the αII structure as an anode material for lithium-ion batteries. VOPO4·2H2O was prepared via a facile hydrothermal method. Electrochemical prelithiation improved the long-cycle electrochemical performance of VOPO4·2H2O. Furthermore, as lithium ions could easily intercalate the structure of VOPO4·2H2O, it was found to be a promising anode material for Li-ion batteries.
2. Experimental
The VOPO4·2H2O anode material was prepared via a facile hydrothermal method using a previously reported chemical deposition method [8]. Briefly, 1.4 g of V2O5 powder (Aldrich-Sigma) and 7.4 mL of concentrated H3PO4 (Aldrich-Sigma) were added successively to 34.6 mL of deionized H2O, and the resulting mixture was stirred for 2 h. Then, the mixed solution was transferred into a 50-mL Teflon vessel, sealed in a stainless-steel autoclave and heated at 100°C for 8 h, followed by cooling to room temperature. The resulting bright-yellow product was collected by washing the mixture with deionized water and acetone and was then dried at 60°C for 24 h to obtain the final product.
The crystalline structure of the obtained VOPO4 ·2H2O material was examined by carrying out its X-ray diffraction (XRD) analysis using a Rigaku SmartLab diffractometer with Cu Kα radiation (40 kV, 250 mA) over the 2θ range of 10-80o at a scan rate of 0.03o s−1. The surface morphology of the material was examined using field-emission scanning electron microscopy (FE-SEM, JSM 7800F) with energy-dispersive X-ray spectroscopy (EDS). The thermogravimetric (TG) analysis of the VOPO4·2H2O material was conducted on an N-1000 TG analyzer (SCINCO) under an air atmosphere by heating it from 25 to 200°C at a heating rate of 5°C min−1.
The VOPO4·2H2O powder, Super-C65 (Imerys), and polyvinylidene fluoride binder (Kynar) were mixed together at a weight ratio of 80:10:10 in N-methyl pyrrolidone (NMP, Sigma-Aldrich) using an automatic mixing machine (KM tech) to obtain a uniformly mixed slurry for the working composite electrode. The mixture was cast onto a copper foil using a bar coater and a doctor blade and was then placed in a convection oven at 120°C for 10 min to eliminate the NMP solvent from the cast slurry, which was then roll-pressed to ensure inter-particle contact.
The electrochemical performance of the VOPO4 ·2H2O was evaluated using a CR2032 coin half-cell composed of a VOPO4·2H2O anode as the working electrode, a lithium foil as the counter electrode, and polypropylene as the separator (Celgard 2400) in an argon-filled glovebox. The organic electrolyte consisted of 1 M LiPF6 in a mixture of non-aqueous solvents ethylene carbonate (EC) and diethyl carbonate (DEC) (1:1 v/v, Panaxetec) and 5 wt% fluoroethylene carbonate (FEC). Electrochemical tests were performed at room temperature and elevated temperatures. Galvanostatic discharge/charge tests were carried out on a LANHE CT 2001A battery test system within the voltage range of 0.01-2.0 V (vs. Li+/ Li). Electrochemical impedance spectroscopy (EIS) measurements were performed on a multichannel electrochemical workstation (ZIVELAB, MP1, WonATech) at frequencies ranging from 100 kHz to 10 mHz with a voltage amplitude of 5 mV.
Electrochemical prelithiation of VOPO4·2H2O material was carried out using constant current charging with a 0.1 C-rate to a cut-off potential of 10 mV (vs. Li/Li+) in a half-cell with counter and reference electrodes as lithium metal. Then, after prelithiation step, these half-cells were disassembled inside a glovebox and new half-cells were fabricated with prelitiation of VOPO4·2H2O material and lithium metal anode, separator, and fresh organic electrolyte for the following electrochemical measurements described above.
To determine the chemical composition of the electrolyte (1 M LiPF6 EC DEC(1:1 v/v) with 5 wt% FEC) after 50 cycles, the V concentrations of VOPO4·2H2O before and after the prelithiation step were measured using inductively coupled plasma optical emission spectrometry (ICP-OES) (Thermo Scientific iCAP 7000, series).
3. Results and Discussion
Vanadyl phosphates are considered promising active anode and cathode materials for lithium-ion batteries. These materials have seven phases (αl-, αll-, β-, δ-, ɛ-, ω-, and γ-VOPO4) with different structures. For example, the β- and ɛ-phases show an open-tunnel structure, whereas the αl-, αll-, δ-, ω-, and γ-phases exhibit layered structures [10]. Among these phases, the αll-VOPO4 phase with a layered structure and good crystallinity endows the vanadyl phosphate with high theoretical capacity (827 mAh g−1) for anode applications owing to the presence of three-dimensional space consisting of VO6 octahedral and PO4 tetrahedral units, which contribute to the stabilization of the structure during the electrochemical process of the intercalation/deintercalation of lithium-ion sources [16-18]. Fig. 1a shows the XRD pattern of the prepared VOPO4·2H2O, indicating the presence of the αll-phase with a layered structure (JCPDS 36-1472). This indicates that the sample showed no structural changes and impurities after the hydrothermal process. It should be noted that the hydrothermal method is an efficient approach for the synthesis of lithium-ion battery anode and cathode materials with high crystallinity, high purity, good particle size distribution, and stable electrochemical performance. Moreover, this method is cost-effective and offers scalability [19]. The as-synthesized sample was examined by SEM, as shown in Fig. 1b. The sample consisted of smooth nanosheets with rough surfaces. The H2O content of the VOPO4 structure was examined by carrying out TG analysis. The VOPO4·2H2O sample exhibited a weight loss of 17.99%, corresponding to approximately 2 mol of H2O per formula (corresponding to two dehydration steps). This is consistent with the XRD results (Fig. 1c) and the electrochemical stability of VOPO4·2H2O, as shown in Fig. 3. In addition, the EDS results showed the presence of V and P in the sample, confirming the homogeneous distribution of the elements without impurity phases, as shown in Fig. 2.
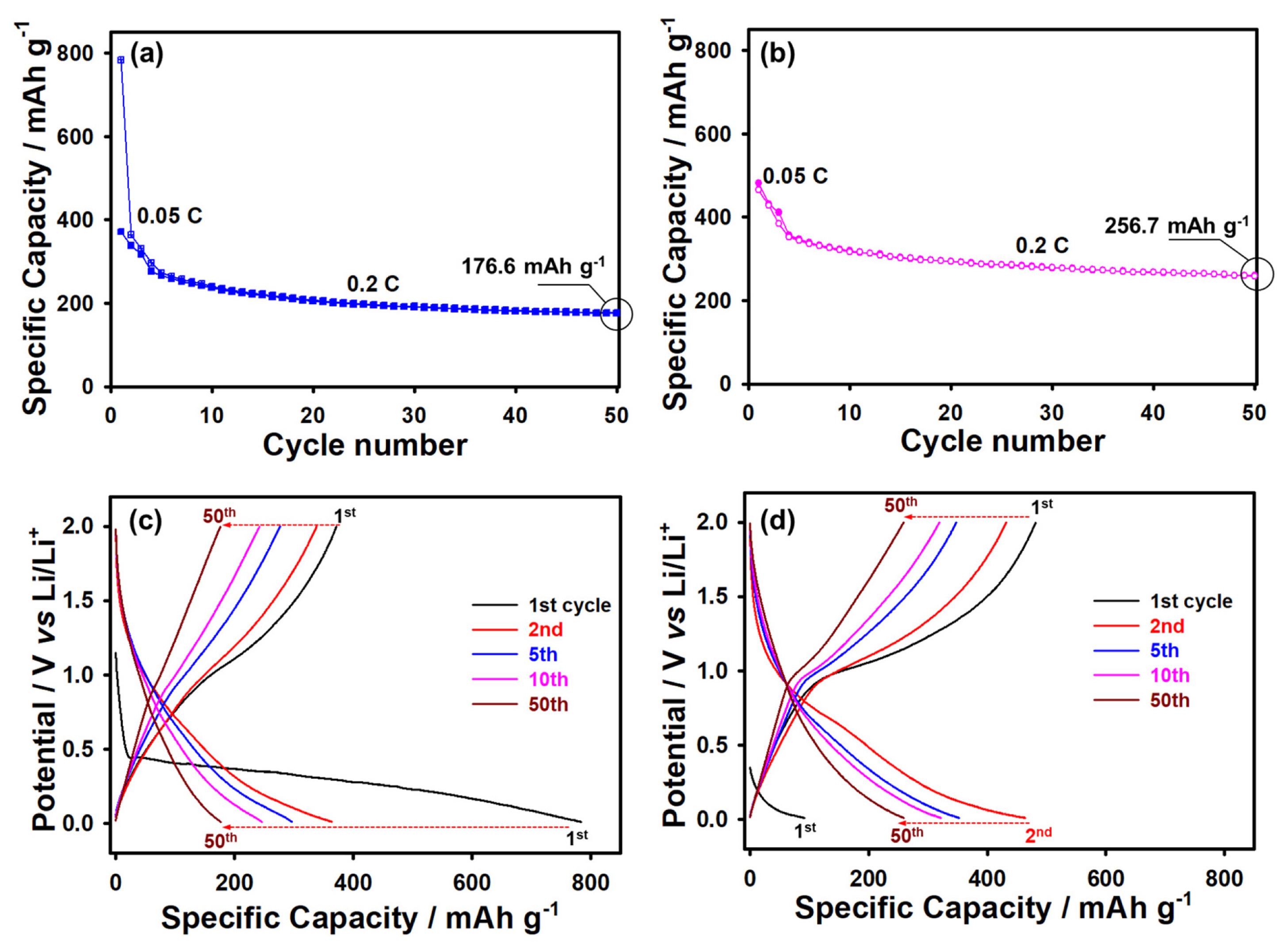
To better understand the redox processes of VOPO4, galvanostatic discharge/charge tests were performed, as shown in Fig. 3a. It should be noted that cycling affected the electrochemical process of the intercalation/deintercalation of Li ions. This is because the V3+/V4+ redox couple for the 1st cycle was not perfectly reversible. This phenomenon is similar to the previously reported conversion mechanism [20-22]. Therefore, the initial specific capacities of the VOPO4 material were 371.5 for de-lithiation and 783.7 mAh g−1 for lithiation. The capacities obtained after the 1st cycle were highly irreversible because of the V3+/V4+ redox couple. The material maintained a specific capacity of approximately 170 mAh g−1 after 100 cycles. This is consistent with the cycling performance and voltage curves of the material (Figs. 3a and 3b) and the results reported previously [10]. In addition, LixVOPO4 was formed during the subsequent charge/discharge cycles (Fig. 3a). Despite the irreversible changes between the V3+/V4+ redox couple, the VOPO4·2H2O sample with the αII structure showed stable cycling at various current densities owing its high crystallinity and structural stability, as shown in Fig. 3d. Another reason is that during the cycling, a solid electrolyte interphase (SEI) layer was formed, which led to a decrease in the specific capacity of the sample and a low Coulombic efficiency of 47.4% after the 1st cycle. However, this resulted in the stabilization of the cycling performance of the material (with a Coulombic efficiency of 99.9%) after 100 cycles [23]. To confirm the electrochemical behavior of the SEI layer, the electrochemical impedance spectroscopy analysis of the VOPO4·2H2O electrodes was carried out during cycling (Fig. 3c). The electrodes showed the electrolyte resistance (Re), capacitance (CPEsei) of the surface film, resistance (Rsei) of the surface film, double-layer capacitance (CPEdl), charge-transfer resistance (Rct), and Warburg (W) impedance related to the diffusion of lithium ions. It should be noted that the resistance related to the formation of the SEI layer increased after the 1st cycle. This indicates that the SEI layer successfully affected the kinetics of the electrode process, thus improving the long-term cycling performance of the electrode (Fig. 3a).
The chemical coefficient diffusion values of the electrodes were determined to evaluate their lithium transport properties at room temperature. These values were obtained from their electrochemical impedance spectra after the 1st and 5th cycles for each temperature and electrolyte. The diffusivity of the sample (DLi) was estimated using Equation (1) [24].
where R is the gas constant, T is temperature, A is the area of the electrode, F is the Faraday constant, C is the molarity of the lithium-ion, and u is the Warburg term obtained from the slope of angular velocity and Z’ of the electrode. VOPO4·2H2O showed the chemical diffusion coefficients of 2.87 × 10−15 cm2 s−2 after the 1st cycle and 2.68 × 10−16 cm2 s−2 after the 5th cycle, indicating that it maintained good lithium conductivity during cycling, which contributed to its improved rate capability owing to its excellent transport associated with the formation of stable SEI layer on its surface.
It should be noted that the rate capability is an important parameter affecting the diffusion of lithium from/into the particles of a Li-ion battery anode. Moreover, it increases the contact between the active material and electrolyte, facilitating the rapid transport of lithium ions with small charge transfer resistance. An efficient approach to improve the electrochemical performance of anode and cathode materials is the prelithiation method (chemical and electrochemical), which significantly increases the cycling life of the active material [25-29]. Therefore, the anode prepared in this study was subjected to prelithiation prior to its electrochemical analysis. We believe that this method can compensate for the loss of active Li during the first cycle, reducing the capacity of the anode, as shown in Fig. 4a. The anode subjected to the prelithiation process showed a high specific capacity of 256.7 mAh g−1 as compared to that without prelithiation (176.6 mAh g−1) over 50 cycles, as shown in Fig. 4b. It should be noted that the prelithiated VOPO4 sample could maintain high and stable capacities as compared to the sample without prelithiation because of its lower resistance (i.e., high Li-ion conductivity and negligible electronic conductivity) and higher lithium-ion selectivity and permeability (Figs. 4a and 4b). In addition, this finding can be attributed to the fact that in the absence of the prelithiation step, an SEI layer was formed on the surface of the VOPO4 anode, which increased the surface resistance and suppressed the diffusion of lithium between the interface of the electrolyte and electrode. On the other hand, the prelithiation step suppressed the dissolution of vanadium ions and the formation of unfavorable side reaction components. Therefore, the unlithiated sample showed higher vanadium ion concentration (1.5610 ppm) than the prelithiated sample (0.7874 ppm) in 1 M LiPF6 EC DEC (1:1 v/v) with 5 wt% FEC after 50 cycles (Fig. 4), as revealed by the ICP-OES analysis results. These results indicate that the VOPO4·2H2O sample exhibited poor performance owing to the sluggish kinetics and possible formation of side reaction components and the dissolution of vanadium ions, which are typical of vanadium-based materials [30]. The cycling potential did not affect the crystalline lattice of VOPO4·2H2O (with the αII structure) and the behavior of the V3+/ V4+ redox couple. However, it was possible to form various Vx+/Vx+ redox couples at potentials higher than 2.0 V, as reported in [31]. It can be concluded that the prelithiation step improved the cycling performance of the anode material. Moreover, the LixVOPO4·2H2O phase formed during the prelithiation step further improved the performance of the anode material.
On the basis of these results, it can be concluded that the VOPO4·2H2O nanosheets with the αII structure obtained via a facile hydrothermal method exhibited stable cycling performance as an anode material for lithium-ion batteries. Furthermore, various phases of VOPO4 materials, are promising anode and cathode materials for Li-, Na-, Mg, Zn, and K-based storage systems and supercapacitors as well as coating materials for high-energy density cathode materials (Table 1).
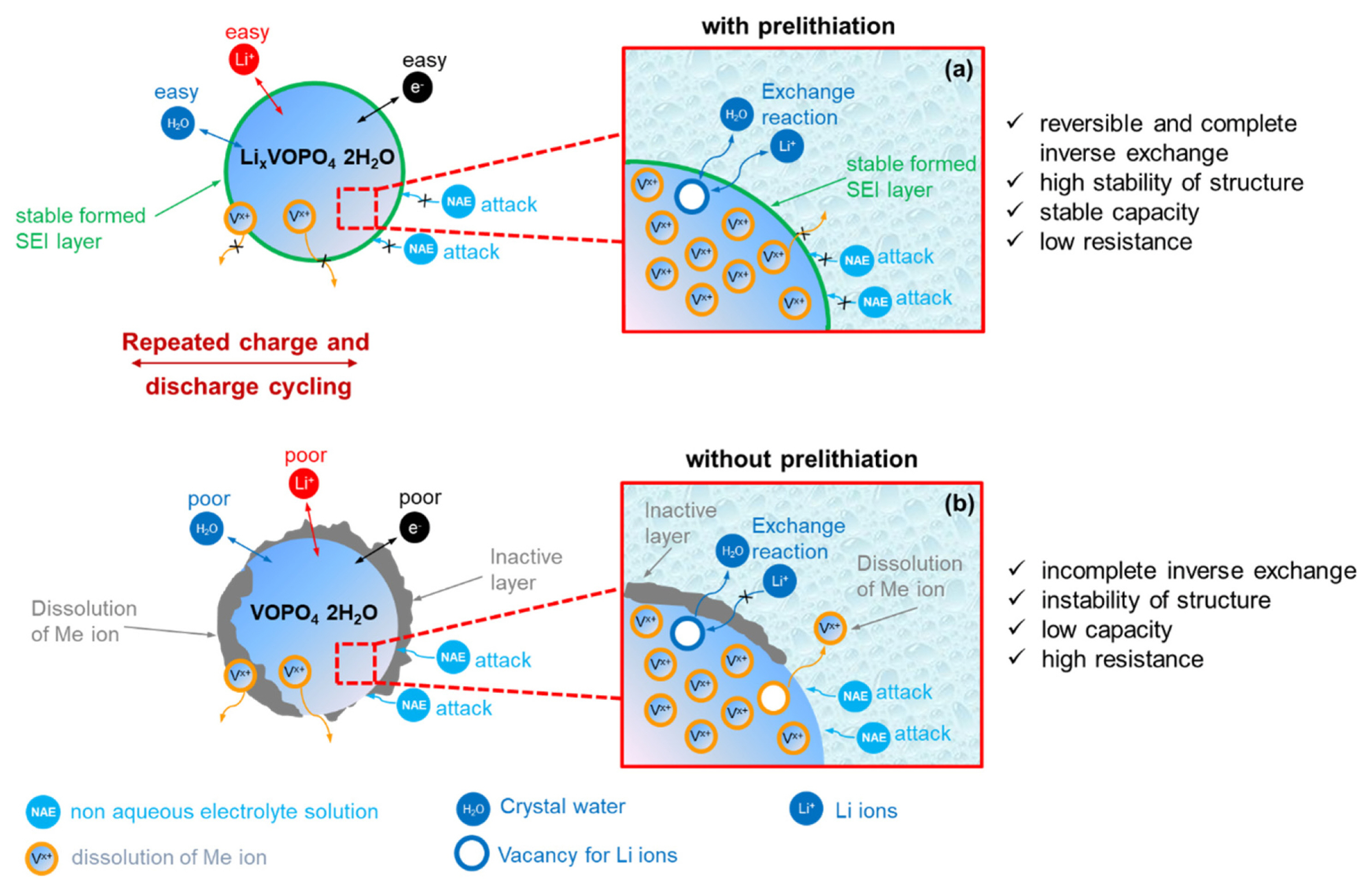
On the basis of the physicochemical and electrochemical analyses results, we proposed a mechanism for the electrochemical exchange reaction between lithium ions and the crystal water molecules of VOPO4·2H2O in the organic electrolyte during repeated charge and discharge cycling affecting the cycle life of the material, as shown in Figs. 5 and S1 (Supplementary Information). First, it should be noted that vanadyl phosphate-based materials with various structures have been investigated as anode and cathode materials for diverse battery systems (Table 1) [38-40]. In 1981, Trenze investigated the crystal structure of VOPO4·2H2O [42], which consists of VO6 octahedra and PO4 tetrahedra, forming a tetragonal two-dimensional slab in the ab-plane. The large interlayer space is occupied by crystal water. PO4 tetrahedra is corner-shared with a vanadium atom of the adjacent VO6 octahedron, and the other two atoms of oxygen are dangling oxygen and water oxygen of “structural water”, as shown in Fig. S1 (Supplementary Information). On the basis of the results obtained in previous studies (Table 1) and the crystal structure of the VOPO4·2H2O sample prepared in this study, it can be stated that the structural and crystal water molecules, which exist in the VO6 octahedra and the interlayer of the PO4 tetrahedra, respectively, can significantly affect the structural stability and electrochemical performance of VOPO4·2H2O, regardless of the type of the migrating ion, such a s, Li, Mg, Na, K, and Zn ions [10,12,14,38,40]. Therefore, we believe that an exchange reaction between the migrating ions and crystal water results in the electrochemical intercalation/ deintercalation process in the VOPO4·2H2O structure. The schematics for the occurrence of failure in VOPO4·2H2O and the improvement in its electrochemical performance in 1 M LiPF6 EC DEC (1:1 v/v) with 5 wt% FEC before and after the prelithiation step during repeated charge-discharge cycling are shown in Figs. 5 and S1, respectively. During the discharge process, the lithium ions interacted with the VOPO4 structure instead of the crystal water extracted from the structure. During the charging process, a complete reverse reaction between the Li ions and crystal water improved the stability of the structure and the cycle life of VOPO4 significantly. Thus, the presence of an optimum amount of water in the VOPO4 structure contributed to the excellent electrochemical performance, low surface resistance, and enhanced lithium-ion intercalation and deintercalation kinetics of VOPO4·2H2O (Fig. 4a). Furthermore, the prelithiation of VOPO4·2H2O in the organic electrolyte resulted in the formation of a stable SEI layer on the surface of the active material, which resulted in its stable electrochemical performance. However, in the absence of the prelithiation step, the specific capacity of the material decreased and the surface resistance increased (Fig. 4). This effect was detrimental to the reversible specific capacity of the material and prevents the complete access to the low potential plateau with a low chemical coefficient diffusion compared to the organic electrolyte. Moreover, during the charge and discharge processes, a complete reverse reaction occurred between the Li ions and crystal water. However, in the absence of the prelithiation step, the crystal water molecules reacted with the components of the electrolyte, leading to the formation of side reaction components on the surface of VOPO4 and an insulating layer with poor conductivity, which prevented the insertion and extraction of lithium ions. This is probably related to the amount of dissolved vanadium ions, which affected the molecules of crystal water and had detrimental effects on the electrochemical performance of the material, resulting in the formation of vacancies that could not be filled by the crystal water molecules in the structure, unlike the case of the prelithiated sample. Therefore, in the absence of the prelithiation step, the “crystal water” in the electrolyte reacted with the components of the electrolyte with the formation of an inactive layer, resulting in structural instability and a decrease in the specific capacity of the material due to the ionic and electronic transfer.
4. Conclusion
In summary, VOPO4·2H2O with the αII structure was successfully synthesized via a facile hydrothermal method. The synthesized sample with a nanosheet morphology was used as an anode material for lithium-ion batteries. The nanosheets showed stable long-term electrochemical performance at various current densities. Moreover, the prelithiation step improved the cycling performance of VOPO4·2H2O and increased its specific capacity from 176.6 to 256.7 mAh g−1. The high crystallinity and nanosheet morphology of VOPO4·2H2O contributed to its structural and electrochemical stability and rendered it a promising anode material for Li-ion batteries.