List of Abbreviations
AOPs
Advanced oxidation processes
A/V
Area/Working volume
BDD
Boron doped diamond
BOPs
Biological Oxidation Processes
EO
Electro-oxidation
EPs
Emerging pollutants
MBR
Membrane Bioreactor
âĒOH
Hydroxyl radicals
POPs
Persistent Organic Pollutants
PAHs
Polycyclic aromatic hydrocarbons
THMs
Trihalomethanes
UASB
Upflow anaerobic sludge blanket
UV
Ultraviolet visible
WWTPs
Wastewater Treatment Plants
1. Introduction
Persistent organic pollutants (POPs) and emerging pollutants (EPs) in water are a serious threat to human health and ecological balance since many of them are toxic, carcinogenic and mutagenic [1,2]. In addition, these pollutants are highly recalcitrant and difficult to remove through Biological Oxidation Processes (BOPs). Ttherefore, conventional wastewater treatment plants (WWTPs) are recognized as the main sources of POPs and EPs in the environment [3â5].
In response, several advanced oxidation processes (AOPs) have been introduced, such as Electro-oxidation (EO), UV, ozonation (O3), Fenton, UV/H2O2, UV/O3, UV/H2O2/O3, and UV/TiO2 [6,7].
Of the above, EO is the most studied process due to its several advantages such as: environmental compatibility, versatility, energy efficacy, amenability to automation, and high efficacy in destroying POPs and EPs in water, wastewater and sludge [2][8]. EO has been studied along with several effluents, most notably: tannery [9], electrical industry [10], coke production [11], dyes [12] and hospitals [13]. However, the EO process has been criticized for: (1) its high operating costs related to electricity consumption, (2) its potential to form by-products more toxic than the originals, (3) some effluents may require the addition of supporting electrolytes which elevate operating costs and (4) short life of electrodes, due to deposits of organic material on their surface (passivation/ fouling) [14,15]. These disadvantages can be reduced if EO is combined with BOPs, which are cheaper to operate and work well with biodegradable organic matter, nutrients and solids (components that interfere with EO performance). In this sense, EO can be combined with BOPs as pre-treatment or post-treatment [2]. Studies from the 1980s already suggested this combination in order to make EO affordable [16]. For instance, Wang et al. [11] reduced the cost of water treatment from coke production from 116 to 64 kWh/kg COD, when coupling a biofilter to an EO process. However, decision making criteria on where to locate EO in a coupled system is not entirely clear.
This review aims at understanding the potential benefits and drawbacks of different systems combining EO and BOPs, either conventional (e.g. activated sludge) or novel technologies (e.g. organic bed biofiltration); and how they affect the formation of by-products. Additionally, it takes a look at the advances in the scale-up process, its challenges and recommendations for future research.
1.1 Content of POPs and EPs in different kind of waters
The concentration of some POPs and EPs in different types of water are shown in Table 1. The presence of these pollutants encompasses all types of matrices, from rivers and lakes to influents and effluents from WWTPs. The concentrations of these contaminants in water vary widely, ranging from a few ng/L to several thousand Ξg/L.
Pawlak et al. [17] found a total concentration of PAHs of 6212 ng/L in a lake located in the Polish Polar Station, Horsund in Svalbard, Norway, being naphthalene the most important one with 5530 ng/L; Chen et al. [20] evaluated 10 PAHs in several rivers in the Hangzhou region, in China, an area with industrial antecedents, and found concentrations of between 0.989 Ξg/L and up to 9.663 Ξg/L; Olayinka et al. [28] determined the total PAHs in the bay of Lagos, Nigeria, which ranged from 46 to 507 Ξg/L with the highest presence of Pyrene (22â92 Ξg/L), fluoranthene (9â140 Ξg/L) and Phenanthrene (3â139 Ξg/L). There is a number of sources of PAHs and their concentration can be due to oil spills, atmospheric deposition, wastewater discharge, soil entrainment, and oil infiltration [28,29]. According to the guidelines of the World Health Organization [30], contamination by PAHs is reached at a concentration of above 50 ng/L as an individual component, which is relatively easy to achieve (Table 1). Other POPs have been even more restrictive with the maximum limit recommendations such as Benzo(a)pyrene, which should not exceed 0.7 Ξg/L in drinking water, because it is the most carcinogenic of all PAHs [30].
This is worrisome because the concentration of Benzo(a)pyrene in wastewater can be as high as 90 Ξg/L. In rivers, values of up to 0.168 Ξg/L have been recorded [20].
1.2 Contribution of biological oxidation process for POPs and EPs removal
The removal of pollutants in a biological process depends mainly on microorganisms, the biodegradability and physicochemical characteristics of the pollutant, temperature, redox conditions, the availability of a substrate, among others [31]. Therefore, removal rates for the same pollutant may greatly vary in literature; even more with recalcitrant ones (Table 2). For example, Biel-Maeso et al. [19] obtained similar efficiencies in a denitrification treatment system for naphthalene, fluorene, anthracene and pyrene with 50, 42, 45 and 54%, respectively. Gao et al. [32] used rot-fungus Pseudotrametes gibbosa to remove anthracene and pyrene, reporting removal efficiencies of 46 and 24%, respectively. Treatment with algae has also proven not to be efficient for EPs removal; de Wilt et al. [33] used inoculated Chlorella sorokiniana in urine and treated wastewater enriched with carbamazepine, trimethoprim and diclofenac, for which they reported a removal of 30, 40 and up to 60%, respectively. Of the above, carbamazepine is a highly stable drug that is difficult to degrade even under activated sludge acclimatization conditions. In this regard, Wang and Wang [34] acclimated activated sludge biomass to different initial carbamazepine concentrations (0.2, 1, 5, 10 and 15 mg/L) and reached a 22.8% removal efficiency (Table 2).
In general, if recalcitrant pollutants are well removed through biological treatments, it is achieved through sorption and not biodegradation [31] (Fig. 1). Therefore, the contribution of BOPs to the removal of recalcitrant pollutants (POPs and EPs) is variable and frequently poor, finding higher concentrations in the effluent than in its corresponding influent, on occasions.
2. Contribution of electro-oxidation process for POPs removal
EO is an alternative to a conventional wastewater treatment to remove the most recalcitrant organic pollutants from complex effluents or to transform them into biodegradable compounds by breaking or fragmenting them until their mineralization or conversion into more easily biodegradable by-products [6,37]. This process occurs in an electrochemical reactor called electrolytic cell, which is composed by a tank, an electrolyte (that could be the same waste-water to be treated), a pair of electrodes, an anode (where the pollutants are expected to be oxidized), a cathode (where commonly heavy metals can be reduced but also organic matter) and a power source (Fig. 2). This process relies on oxidation-reduction reactions that occur in the electrolytic cell to depurate waste effluents [2]. Pollutants can be removed in an EO process following direct and/or indirect oxidation.
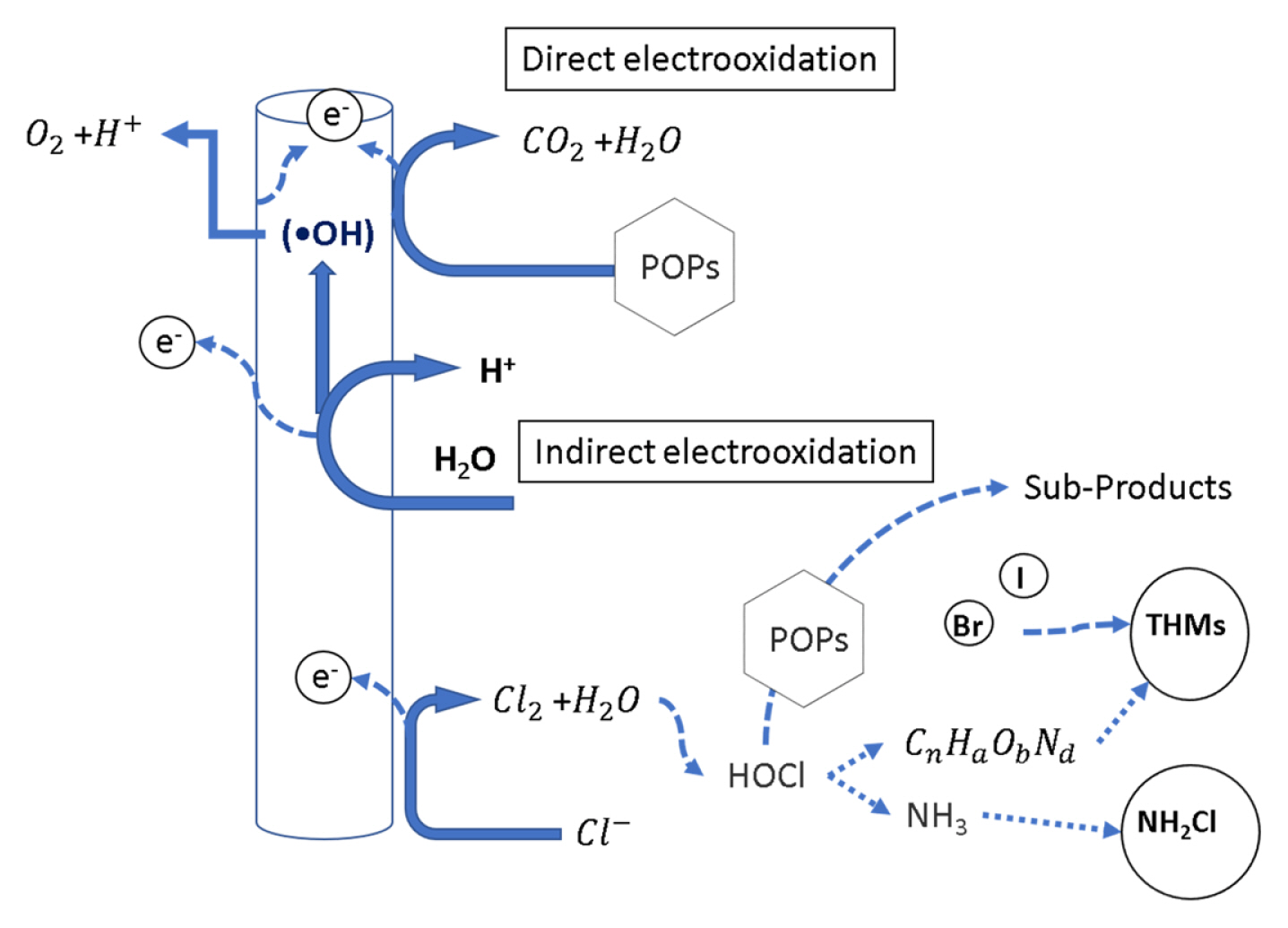
Direct oxidation takes place in two stages: (1) the diffusion of the pollutants from the solution to the anode surface and (2) the oxidation of the pollutants on the surface of the anode by direct electron transfer (Fig. 3). Hydroxyl radicals (âĒOH) are formed by the anodic oxidation of water and physiosorbed at the electrodeâs surface [6,38].
Indirect oxidation occurs when a mediator (HClO, HBrO, H2O2, H2S2O8) is electrogenerated to carry out the oxidation of pollutants [6]. By-products are formed due to the presence of chloride in wastewater which is oxidized into chlorine (Cl2) at the anode. Then chlorine reacts with water to form hypochlorous acid (HClO). This compound can react with organic matter and amines to produce trihalomethanes (THMs) and chloramines, respectively (Fig. 3). The resulting synergy could be highly effective in the degradation of many pollutants. Nevertheless, it has the drawback of toxicity (discussed later) [38].
Side reactions could also occur when the current density applied to the EO process is high enough to overcome the oxidation potential of the anode. In this case, oxygen will be produced instead of âĒOH, thus decreasing the efficacy of the oxidation process. To avoid this, ânon-activeâ electrodes (most commonly BDD and PbO2) are required to generate the hydroxyl radicals (called âactive oxygenâ physiosorbed), which in turn assist with the non-selective oxidation of organic compounds, resulting in their total oxidation to CO2 [38,39]. The electrodes considered âActiveâ such as Pt, IrO2, RuO2, SnO2 and SbO2 show low levels of mineralization attributed mainly to a highly active behavior, i.e., low production of âĒOH [14,40,41].
Due to the simplicity of EO, its application has been evaluated in all kinds of industrial effluents such as: tannery [9], electrical industry [10], coke production [42], dyes [12], landfill leachate [39], olive pomace leachate [43]; and also for other purposes, e.g. disinfection of urban stormwater and secondary effluents [44â46], removal of EPs from hospital effluents [13], among others.
2.1 Advantages and limitations of EO as a single treatment system
Among all AOPs, EO is the most frequently used to remove organic pollutants from a wide diversity of effluents [47]. There is an extensive list of literature related to EO and its application in complex effluents [48â51], and there is also a generalized consensus on their following advantages:
Short retention time. It is possible to apply EOP over short periods of time, mostly of between 60 to 120 min, which translate into small space requirements. However, retention time depends on several factors, namely: complexity of the effluent, water quality requirements, current density/intensity applied and the material of the electrode [8,52,53].
No chemicals
The only consumable is electricity, i.e., it does not require equipment for the addition of chemicals [6,14].
However, EO shows disadvantages when compared to conventional biological processes, such as: the relative high cost associated with energy consumption and the generation of dangerous by-products [54]. Additionally, other possible limitations for EO will be covered along this section.
Although there are several reasons to choose EO as the main treatment to remove recalcitrant pollutants, there are also several factors that must be taken into account which affect its performance:
pH
It is an important parameter that affects the indirect oxidation mainly. In the reactions mediated by chlorine (Clâ), the pH value will affect the proportion of hypochlorous acid (HClO) and hypochlorite (ClOâ). Acidic conditions would be desired since chlorine gas (Cl2) is the strongest oxidant followed by HClO and ClOâ.
Side reactions. The formation of âĒOH occurs at potentials well below the onset of oxygen evolution. The formation of oxygen instead of âĒOH depends mainly on the electrode material which has a specific oxygen evolution potential. If this is high, the formation of âĒOH increases and the formation of oxygen decreases. Oxygen formation, a side or secondary reaction, ends in low current efficiencies for complete mineralization [38], since âĒOH transforms into oxygen before interacting with any other organic molecule (equation 1 and 2):
Passivation or corrosion of the electrodes
It is one of the main problems of the EO in wastewater treatment. Fouling of the electrodes is caused by oligomeric or polymeric material deposited on the surface of the electrode, which causes its deactivation and affects durability [38,55]. Passivation is a type of impermeable layer that builds on the surface of the electrode during its interaction with the pollutant. This layer decreases the transfer rate of electrons between the pollutants and the anode surface and therefore limits the efficiency of the process. For example, Liu et al. [56] investigated the electrode fouling process during EO for water spike with phenol (2 mmol/L). The polymeric layer decreased the electrochemically active surface area of the electrode from 8.38 cm2 to 1.57 cm2 and was developed in barely 100 min of electrolysis (2.0 mmol/L phenol in 0.1 mol/L NaCl at 1.0 V vs SHE).
However, some studies agree that the best anti-fouling strategy is to oxidize the polymeric layer by applying high potentials close to the water discharge region [56â58]. At anode potentials above 2.7 V vs SHE, anions such as chlorine can mitigate electrode fouling by preventing the formation of the polymer layer by active chlorine (âĒ Cl and Cl2) [56].
For instance, Panizza et al. [58] when treating naphthalene sulfonates, found that working with high potentials removed the polymeric film from the anode.
Unfortunately, the higher the current, the greater the deterioration of the electrodes [14,59]. This is especially worrying for electrodes such as PbO2 since, despite being very efficient and relatively cheap to manufacture, they are also not very stable [40] are short-lived and its application may be limited by a possible release of toxic lead ions [48].
High treatment costs
It has been repeatedly proven that EO is not currently applicable at a full scale due to its high implementation cost at pilot and full scales; costly manufacture of electrodes, high consumption of electrical energy by mass of organic matter removed and, at a pilot scale, hydrogen generation makes the operation of the EO complex [39].
Mineralization
Another reason for using EO is the attractive mineralization that is achieved by subjecting the pollutants in close contact with âĒOH and other electro-generated oxidative species. However, it could take a long time to almost mineralize the organic compound. Giraldo et al. [60] could not mineralize oxacillin even after a long exposure time (8 h). In addition, in the vast majority of studies, a complete mineralization is never achieved as revealed by the measurement of total organic carbon or by the detection of by-products of the concern pollutant.
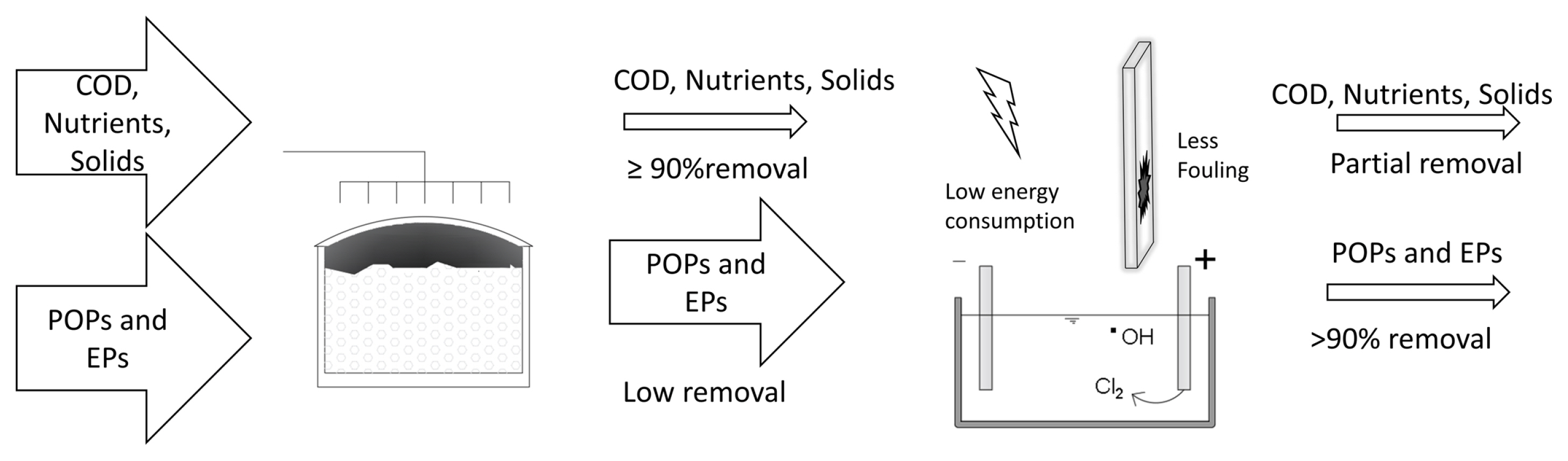
The use of EO as pre-treatment involves dealing with a raw effluent, which could contain high amounts of organic matter, solids, color, turbidity, toxic compounds, among others. All of these factors being able to affect the performance of the EO in the degradation process (Fig. 3).
3. Coupling EO with BOPs
The EOâs main advantage, over other conventional wastewater treatment processes is the conversion of organic compounds into simpler ones and, in theory, up to a final mineralization to CO2 and H2O, which would make the addition of a following treatment module unnecessary. In addition, EO (or any other AOPs) results in the only alternative when the BOPs are not able to deliver the desired quality effluent, e.g., in the treatment of leachates [39]. However, since industrial and/or domestic wastewater effluents are often complex matrices that require long retention times and therefore high energy consumption, in the EO process, in order to reach mineralization levels, its widespread use in real scenarios is greatly restricted [2]. It has been concluded then that if this technology is not complemented with a renewable energy source, it is unlikely to be applied as a sole treatment [41]. Nevertheless, there is an alternative: the coupling of EO with BOPs (Table 3) with the main objective of combining the biological degradation of the organic matter with the potential of the EO to oxidize POPs and to degrade organic matter hardly biodegradable until its mineralization in relatively short times [14] (Fig. 4). As already mentioned in the introduction, this option was proposed since the 80s, but now this line of research has gained interest because the regulatory guidelines are becoming stricter; the pressure on water resources is increasing and the vigilance of the authorities to enforce discharge limits is growing. The following section examines and discusses the advantages and disadvantages of coupling an EO process with a biological treatment, both as a pre-treatment and post-treatment, as well as the role of the oxidizing species such as those derived from chlorine.
3.1 EO as pre-treatment
3.1.1 Electrogenerated by-products
Garcia-Segura et al. [2] says that the configuration of a hybrid electrochemical/biological system will depend mainly on the amount of chlorine and other oxidizing species and the requirements of the treated water.
As mentioned before, indirect oxidation involves the generation of by-products from chlorine (equation 3â6) whose presence in EO is one of the most significant factors (along with electrolysis time and current) in the removal of pollutants; the higher the concentration of Clâ ions, the greater the efficiency in the removal of organic matter and POPs/EPs. Of course there is an optimum that can be found as a ratio COD/[Clâ] in which the removal of organic compounds will no longer increase [37,67].
The presence of Clâ improves the removal of organic compounds and has been evaluated by many authors. Tavares et al. [67] used NaCl (0.2 mol/L) as supporting electrolyte to degrade basic blue 99 dye in 5 min with DSAÂŪ electrodes, unlike the 60 min it took using Na2SO4 (0.2 mol/L) as supporting electrolyte. Similarly, Serrano-Torres et al. [68] degraded Diazo dye Congo (99% removal) faster (5 min) using NaCl (0.05 mol/L) compared to HClO4, Na2SO4 and H2SO4. Differences were significant with Na2SO4 which removed 97% in 180 min. In EPs like pharmaceuticals, Giraldo et al. [60] studied the total transformation of oxacillin to biodegradable compounds in 4 min using 225 mM of NaCl at 30.25 mA/cm2 and Ti/ IrO2 as an anode. This shows how the electrolytic support is a determining factor in the removal of recalcitrant pollutants in conjunction with the current density and the electrolysis time.
However, the main drawback of using NaCl as a supporting electrolyte is the formation of disinfection by-products (DBPs) as a result of the oxidation of organic compounds present in the water. Among these DBPs, THMs are a majority group that include chloroform (CHCl3), bromodichloromethane (CHCl2Br) and bromoform (CHBr3), all possibly carcinogenic to humans [44,69]. On the other hand, with short retention times as those mentioned above, it is very likely that the formation of chlorinated and quinones species continues in the effluent, even at low concentrations. These substances are toxic and biorecalcitrant [49]. In addition, it is necessary to take into account the by-products of the pollutant. For example, Torres et al. [49] completely degraded 5-amino-6-methyl-2-benzimidazolone in 45 min of electrolysis time (Anode: Pt and 50 mA/cm2). However, it resulted in a toxicity increase. After only 4 h of electrolysis, the effluent was considered suitable for biological degradation, which was achieved with a fixed bed reactor. In this regard, Grafias et al. [65] evaluated the treatment of the olive oil extraction agroindustry; using EO with a BDD anode for 360 min and 20 A of current as the first stage of the treatment and then a constructed wetland (CW) as post-treatment (EO/CW). These authors found that the microbial consortium in the CW was negatively affected by the rise of toxicity of the EO effluent, which decreased the removal of COD and color. GarcÃa-espinoza et al. [69] degraded carbamazepine with a BDD electrode and with 14 mM of NaCl as a supporting electrolyte. However, the accumulation of chlorine was reported at longer electrolysis times, which resulted in a greater toxicity for Vibrio fischeri.
There are guidelines for drinking water regarding the discharge of these DBPs. The maximum permitted concentrations for total THMs established by are the USEPA and the European Union 80 Ξg/L and 100 Ξg/L, respectively [70]. These limits could be easily exceeded, for example, PÃĐrez et al. [70] treated the rejection of a reverse osmosis process (with 800 mg/L of chloride) and found that by applying 20 mA/cm2 almost 200 Ξg/L of THMs were present in the treated effluent (exceeding the guidelines). Feng et al. [44] used 110 mg/L of chloride and 4.2 mA/cm2 during 20 min for the disinfection of rainwater and generated 34 Ξg/L of THMs, concentration below the guideline limits but using a relatively low current density and short time.
3.1.2 Enhancing biodegradability
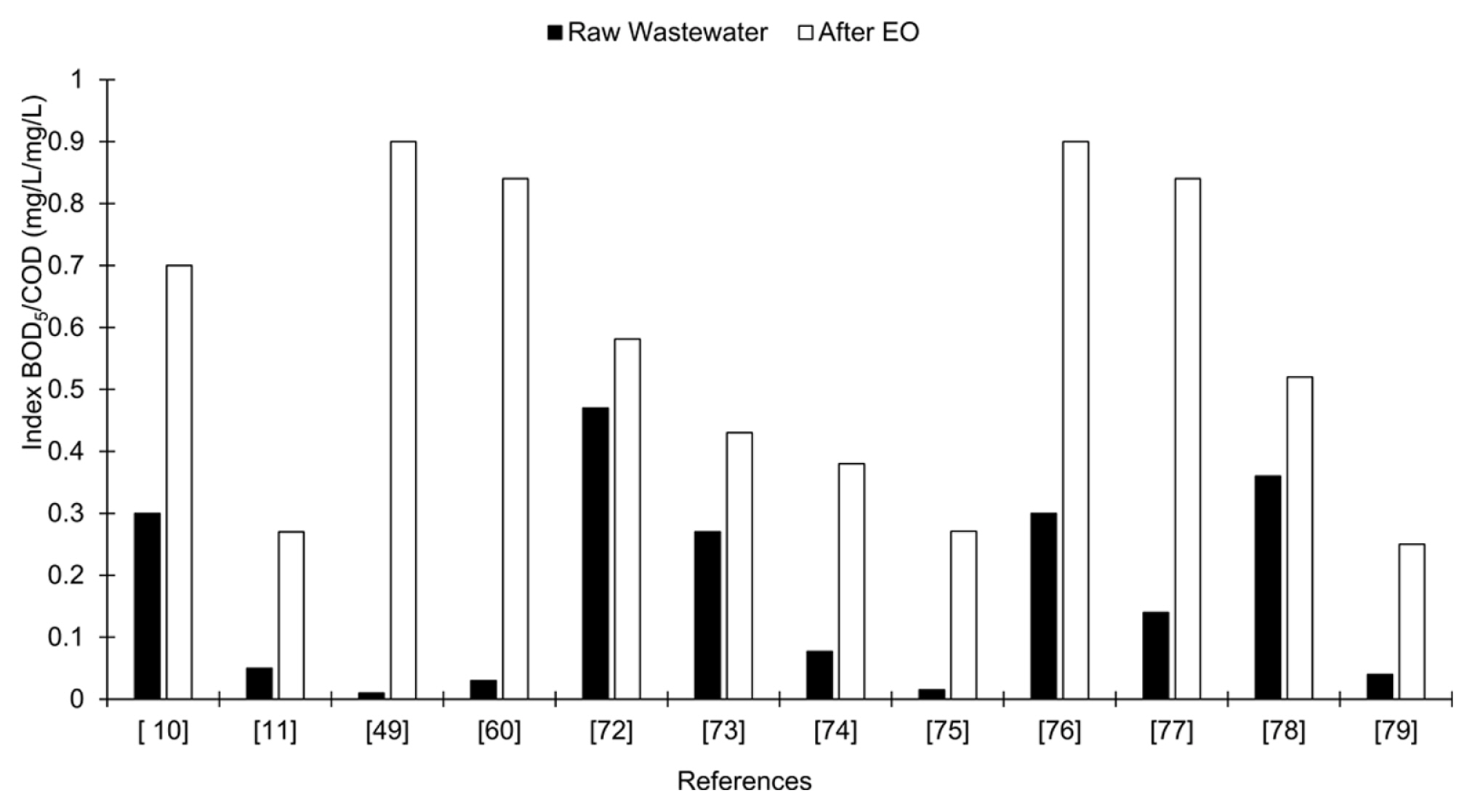
The desirable effect of EO as pre-treatment is to enhance the biodegradability of the industrial effluent to be treated. The aim of the pre-treatment is to partially oxidize the persistent part of the effluent and to produce biodegradable intermediates [15]. The percentage of mineralization, however, must be minimal in order to allow the subsequent BOPs to degrade these intermediaries and thus avoid unnecessary expenditure on chemicals and energy [71]. Examples of works where the biodegradability of the effluent is improved after an EO process, increasing the BOD5/COD ratio are presented in Fig. 5.
A BOD5/COD ratio of 0.4 is considered as the limit of biodegradability to submit an effluent to biological treatment [66]. Therefore, for values below, it is highly advisable to pretreat with EO, as is the case of many industrial effluents such as irrigation wells contaminated with herbicides (2,4-Dichlorophenoxy-acetic acid) paper, dyes, pharmaceutical effluents, leachates, among others.
3.1.3 Interferences by solids
One of the limitations of the EO is that the persistent pollutants of interest must be adsorbed first at the anode in order to be directly oxidized [66]. If the EO is used as a pre-treatment, the solids present in the raw wastewater make it difficult to transfer the recalcitrant pollutants to the anode, reducing the efficiency of the process. Additionally, many POPs and EPs can become adsorbed on solids, which reduces the possibility of them getting in contact with electro-generated âĒOH. Barrios et al. [8] evaluated the performance of an EO process with BDD electrodes to treat sludge with a concentration of 0.8% and concluded that the low degradation of non-phenols and triclosan was due to the fact that >98% of these compounds remained associated (adsorbed) to solids. Efi Kotta [80] found a decrease in the efficiency of the treatment of organic matter due to the presence of high concentrations of solids (80 g/L) in the industrial effluent of olive bleached pulp, in addition to the rise of soluble COD due to the oxidation of solids at the anode. GarcÃa-espinoza et al. [69] found a reduction in the first-order kinetic constant in the degradation of carbamazepine by using demineralized water, tap water and treated residual water of 0.189, 0.071 and 0.0351/min, respectively, which could be explained by a possible competition of solids and carbamazepine for the hydroxyl radicals or, by the fouling of the electrodes that affect the efficiency of the process.
3.2 EO as post-treatment: Advantages and disadvantages
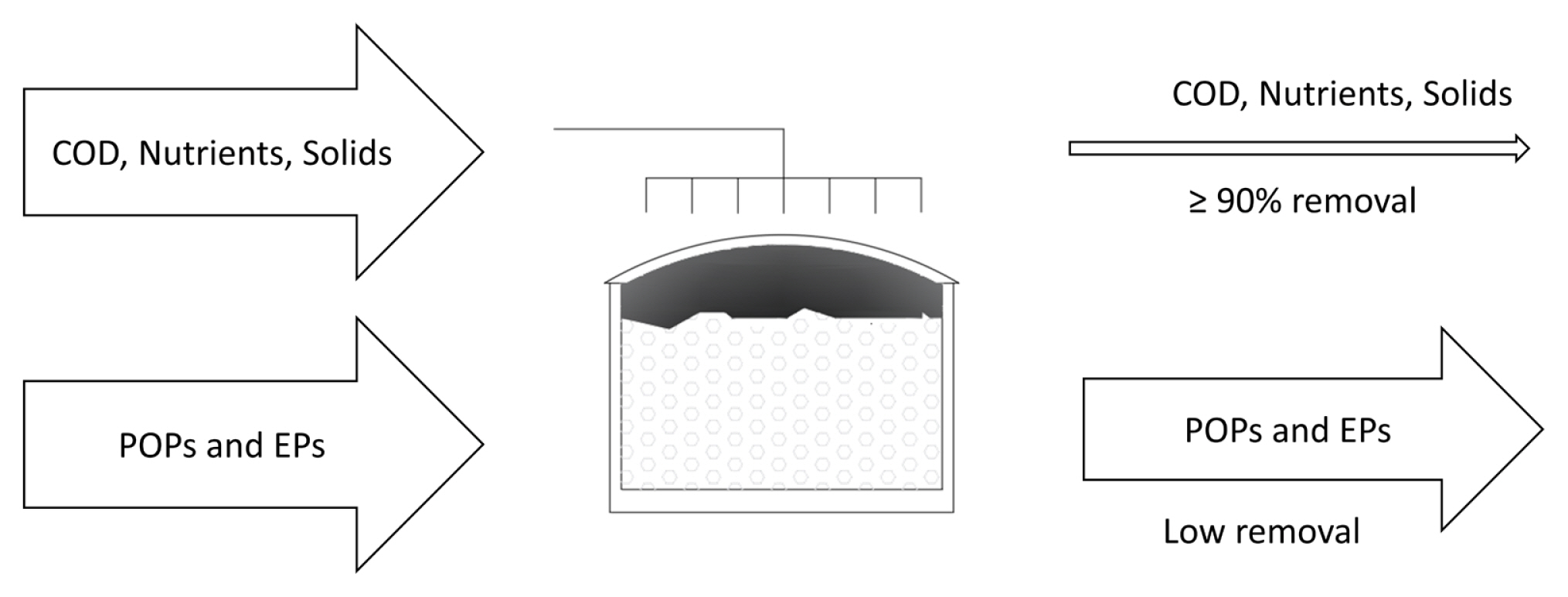
Unlike EO as a pre-treatment, EO as a post-treatment is a less frequent arrangement but there are studies that have been carried out for all types of effluents: industrial, municipal, domestic and even of natural water currents (rivers) (Fig. 6). There are also few reports that compare both treatment configurations, that is, EO as pre and post-treatment and thus evaluate the differences in different types of effluents.
EO as post-treatment takes place in the following cases: (1) after the biodegradable organic pollutants have been previously degraded in BOPs in order to oxidize the remaining organic matter (which is usually more bio-resistant), (2) to remove trace contaminants such as pharmaceuticals and pesticides and (3) to remove color in case of reuse in an industrial process such as paper and textiles. In this way, all kinds of effluents have been studied, such as: traces after an anaerobic treatment Vidal et al. [81]; reverse osmosis rejection treatment that treated water from a secondary effluent [82â84]; leached from olive pulp previously treated in a CW [65] and through an upflow anaerobic sludge blanket (UASB) [43]; leachate from sanitary landfills pretreated with activated sludge [39,61], or pretreated by a membrane bioreactor (MBR) [62] and by ultrafiltration [63]; the treatment of paper pulp pretreated with an UASB [85]; tannery (it does not specify biological process) [86]; door manufacturing processes with activated sludge [87] and even hospitals previously using a MBR [13].
During the EO as a post-treatment, a decrease in the risk unwanted by-products formation such as THMs can be expected. Frontistis et al. [40] for example, found a decline in aromatics content after 30 min of EO, however, risk was not evaluated.
In hospital effluents, EPs like pharmaceuticals are of great interest. These effluents are characterized by concentrations of pharmaceuticals from 4 and up to 150 times higher, compared to domestic and urban [88]. In this sense, Ouarda et al. [13], found that when using a MBR, the removal for carbamazepine and venlafaxine is very limited (<10%), but by submitting the biologically pretreated effluent to EO, it was possible to remove these drugs in 40 min and with 0.5 A (Nb/BDD). On the other hand, when the EO was used as pre-treatment, the removal of carbamazepine and venlafaxine was only 50 and 66%, respectively. After applying a MBR process as a polish, the venlafaxine removal rose up to 92%. This leads to concluding that the EO as a pre-treatment followed by a MBR is not the best option for hospital effluents. However, the configuration of EO as a post-treatment generates a toxic effluent to Daphnia magna and Vibrio fisheri if it is not diluted (100% v/v toxicity). It was concluded that the EO as a post-treatment not only improves the removal efficiency of the evaluated pharmaceuticals but also decreases the energy consumption when going from a current intensity of 2 to 0.5 A.
In this sense, Panizza et al. [58] found that treating an industrial effluent containing naphthalene sulfonates led to achieving mineralization when using EO as the only treatment, consuming 80 kWh/m3 in 4 h. However, by applying a hybrid system biofilter/EO, the energy consumption decreased to 61 kWh/m3 in 3 h. This means that the fouling of the electrodes caused by the increase in the organic load (when EO was used as pre-treatment) forced the electrolytic cell to be operated at a higher current than stoichiometrically required.
Iniesta et al. [57] found that at a low current density, high phenol concentration and low conversion, phenol is oxidized to aromatic compounds (benzoquinone, hydroquinone and catechol) which, according to Liu et al. [56], these type of compounds could further combine with phenol radicals to generate phenoxy phenol or dihydroxyl benzene which then transformed into a coating layer that covers the anodeâs surface. Since biological treatment removed phenol from water, fouling risk decreased at low current densities.
4. EO in pilot scale applications
The background of the EO in terms of wastewater treatment is sufficient to demonstrate the viability of the technology to remove POPs, EPs, organic matter, nutrients and pathogens from all types of waste effluents. Therefore, the logical step to follow is to apply this technology on a larger scale (pilot or full-scale). This section will examine the progress in scaling up EO processes. The methodology details and the problems encountered during the implementation process (for large scale application) will also be presented.
In order to scale conventional biological processes (activated sludge, trickling filters, wetlands, etc.) from results at the laboratory level, it is very important to preserve the dimensional relationships of the corresponding reactor. However, in the case of electrochemical reactors, this criterion usually cannot be met, since the increase in the interelectrode space would result in a high voltage drop and, therefore, an increase in energy costs. Thus it is always better to reduce this distance as much as possible within a certain limit [89,90].
The safest route for the scaling of electrochemical reactors is through the use of multiple laboratoryscale electrochemical cells [91]. According to Anglada et al. [39] and Urtiaga et al. [51] the degradation constant of the pollutant will be similar regardless of the number of cells that are operated simultaneously, while the hydrodynamic conditions are maintained. Thus, the model proposed by Comninellis and Chen. [38] represented in equations 7 and 8 can be applied.
Where jlim is the limiting current density (A/m2), A (m2) is the area of the anode, V (m3) is the volume of treated wastewater, km is the mass transport coefficient in the electrochemical reactor (m/s), COD is the chemical oxygen demand at a given time and F is the Faraday constant (C/mol).
From a series of degradation tests and calculating the mass transport coefficient (km), Anglada et al. [39] scaled up a process from laboratory test up 150 times, that is, with a total BDD anode area of 1.05 m2 and using several individual cells (area per cell 70 cm2), the results obtained for a single cell satisfactorily described what happened at the pilot level, for the removal of both organic matter and ammonia nitrogen. Since the EO is a non-selective process, the kinetic models apply to any type of molecule that adds COD to the system [92]. In this sense Urtiaga et al. [51] carried out a series of degradation kinetics at laboratory scale using different operating conditions (initial COD 1500 â 3000 mg/L, current density 300, 600 and 1200 A/m2) in order to scale up; COD degradation levels reached in the pilot scale validated the procedure in which the A/V ratio was preserved.
There is a shared opinion among some authors that the safest route for scaling is multiplying the number of cells tested at laboratory scale. This is challenged by others who say that it is likely that the scaling can be performed only by maintaining hydrodynamics of the cell studied at the laboratory level and maintaining the A/V ratio. For instance, Zhu et al. [48] scaled a 24 cm2 BDD anode 121 times (2904 cm2) for the removal of phenols. The dimensional relations were maintained in the configuration of the electrode and in the working volume. However, the interelectrode distance and the operating variables (current densities, retention times, conductivity and initial COD) were the same in both scales. It should be noted that the analysis of the kinetic behavior was not performed and only the hydrodynamics and the A/V ratio were maintained. After the scaling up, the results were very similar to those obtained at laboratory scale according to the response surface methodology in which a relatively small standard deviation of 0.2 to 12% was found.
Another way to approach scaling is the one presented by Abou et al. [93], who used a single cell with an effective volume of 19.2 L that contains 49 graphite electrodes with a total area of 0.0126 m2, maintaining a distance of 2 cm for each pair of electrodes (Fig. 7). In this way, a large number of electrodes can be installed until the desired A / V ratio is reached. This pilot prototype was evaluated to remove phenols from wastewater from the oil industry, obtaining > 99% efficiency in phenols and 40 to 60% removal of COD.
More recently, Monteil et al. [94] developed a novel pilot-scale reactor to operate in continuous mode. This reactor consists on a cell with a useful volume of 22.5 L in the form of a channel that works with 14 and 28 pairs of electrodes (BDD) (Fig 8). The authors evaluated the effect of the flow (20.4 to 170 mL/min) on the mineralization of the drug hydrochlorothiazide in the solution. The results showed that the flow rate was the most critical parameter: the lower the flow rate, the higher the mineralization percentage. The number of electrodes in both configurations allowed a high mineralization efficiency (> 90%).
The scaling of EO, although successful, it is currently limited to a pilot scale; the main constraint being that, in order to treat a relatively large effluent, several hundreds of small electrolytic cells are required. The technological development should be focused on the creation of large electrodes (such as those used by Zhu et al. [48]) that result in the use of a small number of electrolytic cells to treat high volumes or be focused on the creation of large electrolytic cells with many electrodes (such as those used by [93,94]). On the other hand, BDD electrodes are considered to deliver the best performance in waste-water treatment. However, it is very expensive when compared to DSAÂŪ, carbon or graphite electrodes [92,95]. Therefore, a larger research is needed using these electrodes at a pilot scale.
5. EO coupling to BOPs challenges and future developments
âĒ Taking into account that DBPs are dangerous substances, all studies related to EO should closely monitor their formation. It takes place while there are low concentrations of Clâ such as 119â144 mg/L, or as high as 4500 mg/L [37,45] and the applied current density is high enough to favor the evolution of Clâ to DBPs.
âĒ There are still insufficient studies that rigorously monitor the formation of DBPs in the final discharge, as well as the optimal COD/Clâ ratios to avoid an increase in the toxicity of effluents treated by EO. There is a limited number of proposals to solve this problem which, along with the high costs, is a great limitation for scaling up.
âĒ There are scarce studies in passivation/fouling focused on long retention times and cleaning/reactivation methods. Efforts are needed in this direction to investigate the feasibility of this technology on a pilot / industrial scale.
âĒ The future developments to scale up the EO process should be aimed at manufacturing cheaper and larger electrodes, in order to decrease the number of electrolytic cells used, thus increasing the treatment capacity to full scale.
6. Conclusions
EO has proven to be an efficient process to remove recalcitrant pollutants. In addition, its implementation is relatively easy and versatile to treat different effluents. However, in general, it is expensive due to two factors: 1) Long reaction times are required, sometimes hours if the mineralization of POPs and EPs is to be achieved, and 2) the manufacture of some electrodes is expensive.
On the other hand, the unavoidable generation of toxic disinfection and degradation by-products complicates the reuse and / or discharge of an EO-treated effluent. Therefore, various studies recommend that EO be used in combination with a biological process. EO as a pre-treatment is generally applied to increase the biodegradability of an industrial effluent. However, the operation is complicated because: 1) Sufficient substrate must be maintained in the effluent for the subsequent biological process and 2) Generally, there is little control regarding the generation of toxic by-products of disinfection and degradation, which affect the microorganisms of the subsequent biological system. In this sense, the most promising configuration of this coupling seems to be EO as a post-treatment, since solids, organic matter and nutrients are eliminated in the biological stage, thus concentrating the generation of OH radicals from EO to mineralize the more persistent pollutants. By applying EO as a post-treatment, the generation of disinfection by-products such as THMs could also decrease by limiting their precursors such as organic matter or nutrients. An additional benefit is to decrease the formation of a polymeric layer on the surface of the electrodes and therefore their fouling.
So far, the number of studies carried out with EO on an industrial / real scale is limited, although the results obtained on a laboratory scale on a pilot / industrial scale have been successfully replicated. This has been achieved by maintaining the hydrodynamics of the electro-oxidation cell (same A / V ratio) either by using many laboratory-scale cells connected in parallel or by developing larger electrooxidation cells with a pair of electrodes that are also larger or by placing many laboratory-scale electrodes distributed in larger electrolyte cells.