1. Introduction
A fuel cell is a device that converts the chemical energy of hydrogen into electrical energy and operates by oxidizing hydrogen at the anode and reducing oxygen at the cathode [1]. Due to the sluggish reaction kinetics of oxygen reduction reaction (ORR) at the cathode, a larger amount of platinum is used in the cathode than in the anode [2]. Therefore, in 2022, the cost of the catalyst occupied about 60% of the stack cost of $112 kWnet â1 in the 275 kWnet proton exchange membrane fuel cell (PEMFC) system for the heavy-duty vehicle (HDV) at 50,000 systems yrâ1 [3]. In 2020, the cost target of the membrane electrode assembly (MEA) for an 80 kWnet PEMFC stack was set at $14 kWnet â1 for transportation applications by the U.S. Department of Energy (U.S. DOE) [4]. Thus, to lower the cost of the stack, it is essential to reduce the catalyst production cost by developing low-precious or non-precious metal catalysts. As an approach to achieving it, anion exchange membrane fuel cell (AEMFC) has recently been attracting attention [5].
AEMFC has a few differences from PEMFC in terms of components and structure. The significant difference is that instead of protons being conducted from anode to cathode through a proton exchange membrane, hydroxyl ions (OHâ) are transferred from cathode to anode through an anion exchange membrane (AEM). First, at the cathode of AEMFC, the reduction of O2 resulted in OHâ ions, and the OHâ ions passed through the AEM. Then, OHâ ions react with hydrogen at the anode to produce water. The generated water is transferred to the cathode by back-diffusion [6,7]. Since ORR kinetics in an alkaline media is faster than in an acidic media, a smaller amount and broader range of catalysts can be used for AEMFC than PEMFC [8,9]. In addition, due to the high pH, the components used in the fuel cell system also have an advantage in more affordable materials can be used because there is less concern about corrosion, and it was expected that non-precious metal catalysts could be applied [10â12].
A Pt/C, a traditional ORR catalyst, would have durability issues, carbon corrosion, and detachment and agglomeration of Pt in alkaline conditions with a still high price of Pt [13,14]. Therefore, interest in non-precious metal catalysts that are relatively stable and inexpensive under alkaline conditions has been increasing [15â17]. Among these, one of the most actively studied structures of the ORR catalytic active sites is M-Nx, in which a transition metal (M) is hosted in a carbon matrix with multiple N coordination (Metal-N-C; MNC) [18,19]. Research on improving the activity of MNC by introducing heteroatom dopants such as S, B, P, or various types of transition metals (e.g., Fe, Co, Cu, etc.) to MNC has been actively conducted [5,20â23].
These attempts have become more specific and advanced by deepening understanding of the evolution pathway of the active sites and mechanism of the ORR catalysis [24,25]. Accordingly, studies have been conducted to improve the ORR kinetics by controlling the d-band center of the active metal or changing the spin state by introducing an axial ligand. For instance, halide anion including Clâ was bound to the M-Nx site as an axial ligand, or coordinated N in M-Nx sites was replaced with a heteroatom to modify the electronic state of the active site [26,27]. However, despite these progresses, the low density of active metal, still limited to several wt.% of the total catalyst mass, and mass transport resistance due to lack of the interface between the active site and reactants are still recognized as issues to achieve high-performing ORR catalysts comparing that of Pt/C [28,29]. Therefore, attempts to increase the density of active sites in the catalyst using sacrificial metals (e.g., Zn) during synthesis or to address problems such as mass transfer resistance by designing a suitable pore structure for ORR are continuing [30â32]. PEMFC also shares these issues and research has recently been conducted on electrochemically stable and dense active sites such as the Fe-pyridinic N4 site and morphology control for improved mass transport, which could be applied to the AEMFC system [33â35].
In this review, the footprints of MNC catalysts for AEMFC cathodes and recent research trends related to active site engineering and morphology control of catalysts will be discussed.
2. Development of MNCs
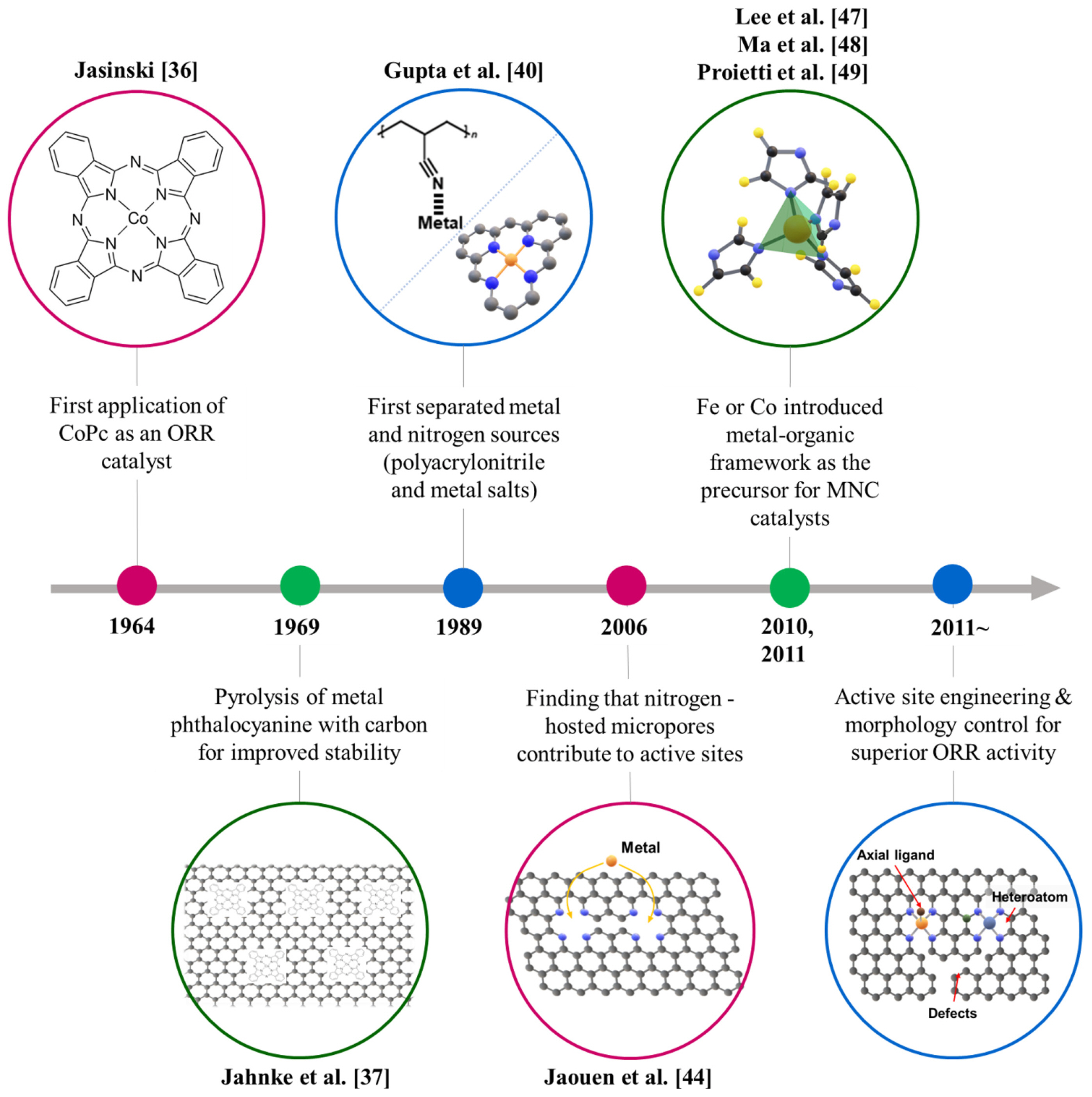
2.1. Development history of MNCs before the 2010s
Since Jasinski suggested the possibility of metal phthalocyanine as an ORR catalyst, there has been discussion about approaches based on nitrogen-containing macrocyclic metal complexes (Fig. 1) [36]. At first, metal phthalocyanine-promoted carbon black was used as an electrode, however, in an acidic environment, the macrocycles decomposed and the center metals were leached out, showing low stability [37,38]. To achieve stable MNCs, heat treatments of macrocyclic metal complexes with carbon black were proposed [39]. In addition, Gupta et al. separated the N precursor and metal source by pyrolysis of polyacrylonitrile with metal salts, enabling the development of a more flexible synthesis method [40]. Based on these efforts, researches on methodologies of forming active sites using metals and N-containing carbon sources such as pyrrole and phenanthroline were conducted [41â43]. However, unlike the development of the synthesis method, the understanding of the evolution mechanism of the active site of pyrolyzed MNC was still insufficient.
Jaouen et al. suggested that M-Nx active sites are hosted in micropores surrounded by nitrogen by observing changes in the physical and electrochemical properties of the catalyst depending on heat treatment conditions [44]. Then, approaches have mainly been attempted for pyrolysis of metal-chelated composites such as polyaniline-metal complexes or forming active sites with a microporous carbon support [45]. In the latter, the metal-organic framework (MOF), which is one of the most frequently used methods for the synthesis of MNCs, was included [46]. MNC using MOF was first reported in 2010 and 2011, the PEMFC performance containing a ZIF-8-based Fe-N-C cathode was reported to have a peak power density of 0.91 W cmâ2 in H2-O2 condition, which accelerated research on MOF-based MNCs [47â49]. Pyrolysis of ZIF-8 and Fe or Co precursors composite fixed Fe or Co ions to the N-hosted micropore in ZIF which was developed by gasification of Zn (b.p. of Zn: 907°C) to form an M-N4 site [31,50].
However, since the agglomeration of metal particles was inevitable during the heat treatment to form and stabilize the metal-Nx sites, it was attempted to suppress the growth of metal particles by adding inhibitors such as silica and chelators to prevent metal particle agglomeration [51â53]. Nevertheless, the amount of catalytically active metals in MNC was limited to several wt.%, and electrochemical behaviors in a single cell were not yet fully understood [5,33]. Therefore, the performance of MNC in a single cell was inferior to that of a Pt catalyst even though the higher activity of MNCs in half-cell than that of Pt/C is achieved in alkaline conditions. To overcome performance limitations due to low metal content, studies on morphology and active sites of MNCs have been conducted to improve ORR kinetics and mass transport [5]. In the next chapter, first, approaches to control the morphology of the catalyst, which attributes mass transport with hierarchical pore structure for facilitated oxygen and charge transfer, were introduced. Then, it presented recent advances in the design of active sites of MNC to improve reaction kinetics by modulation of the electronic state of the active site with heteroatom introduction and defect engineering, which shifts the adsorption energy between the reaction intermediate and the active site.
2.2. Morphology control of MNCs for accessible active sites with high mass transport ability
First of all, in the pore structure of MNC, micropores, mesopores, and macropores have different roles. Micropores are involved in the formation of an active site because Fe is fixed to a defect surrounded by multiple Ns and forms the M-Nx moiety [44]. Mesopore increases the electrolyte-wettable area of the catalyst. Macropores contribute to mass transport such as O2 transport [54]. For highly active FeNC, a hierarchical pore structure where all three pores are developed is ideal. The AEMFC performance using MNC catalysts with such characteristics is summarized in Table 1.
Adabi et al. reported the improved performance of AEMFC with commercial FeNC cathode, peak power density of 2.05 W cmâ2 and current density of 7.4 A cmâ2 @0.1 V (H2/O2 flow) of AEMFC (Cathode: FeNC 1 mg cmâ2; Anode PtRu/C 0.6 mgPtRu cmâ2) using commercial FeNC with hierarchical pore structure from VariporeTM method which uses fumed silica as a hard template [55]. It was achieved by optimizing the operating conditions, backpressure, relative humidity, and temperature of the cell to control the water balance of the MEA. The performance of 0.66 V at 1 A cmâ2, exceeding the 2022 U.S. DOE milestone (0.65 V at 1 A cmâ2), was observed in MEA using a low precious metal anode of 0.125 mgPtRu cmâ2 meeting U.S. DOE target set in 2020 [4]. It was found that when the temperature of the cell and humidified inlet gas was assigned to a relatively low temperature of 60°C, there was a problem of water accumulation in the anode, resulting in swelling of the anode ionomer. It was suppressed by controlling appropriate temperature and humidity conditions, and backpressure was applied to the cathode to remove water inside the cell.
Designing catalyst morphology to have a hollow structure was considered one of the approaches to increase active site utilization. For this purpose, open-mouthed particles of hollow cobalt/nitrogen-doped carbon with mesoporous shells (OMH-Co/NC) were proposed [69]. Zn and Co sulfate were dissolved in methanol to form a metal sulfate sphere, and then 2-methylimidazole was added to coordinate with the metal sulfate on the surface (Fig. 2a). OMH-Co/NC was obtained by heat-treating the formed raspberry-shaped R-Zn/Co-ZIF (Fig. 2b,c). OMH-Co/NC was confirmed to have a mesopore developed in the form of a hollow porous sphere with an opening at one end (Fig. 2bâe). When ORR activity was evaluated in a half-cell (0.1 M KOH), a half-wave potential of 0.865 V and a limiting current density of 5.7 mA cmâ2 at 1600 rpm were obtained (Fig. 2f). The hollow structure and open hole of OMH-Co/NC contributed to low mass transport resistance and charge transfer resistance (Fig. 2g,h).
Persky et al. reported a bioinspired Fe-Cu porphyrrole aerogel catalyst [70]. The authors explained that the catalyst has a high specific surface area and a hierarchical pore structure, giving it advantages in density and mass transport at the active site. FeCl2, 5, 10, 15, 20-(tetra-4-aminophenyl) porphyrin, and Cu(II) 5, 10, 15-tris(4-formylphenyl) corrole were chelated to create a gel and then dried using a critical point dryer. Prepared aerogel was carbonized in an inert gas atmosphere at 800°C to prepare HT-FeCu porphyrrole aerogel. HT-FeCu porphyrrole aerogel showed a half-wave potential of 0.80 V and an onset potential of 0.94 V vs RHE in 0.1 M KOH. An onset potential of 0.97 V and a peak power density after iR-correction performance of 0.51 W cmâ2 was reported in AEMFC. Understanding the origin of ORR catalytic activity of HT-FeCu in detail using DFT calculations, adsorption free energy of OH* can be applied as a descriptor of ORR kinetics. It was reported to have the highest ORR activity when the free energy of OH* is in the range of 0.7â1.0 eV [42]. Based on this, as a result of calculating the active site of HT-FeCu porphyrrole aerogel, FeCu-porphyrrole would have an OH* adsorption free energy of 0.796 eV, which was 0.173 eV higher than 0.623 eV of Fe-porphyrin structure.
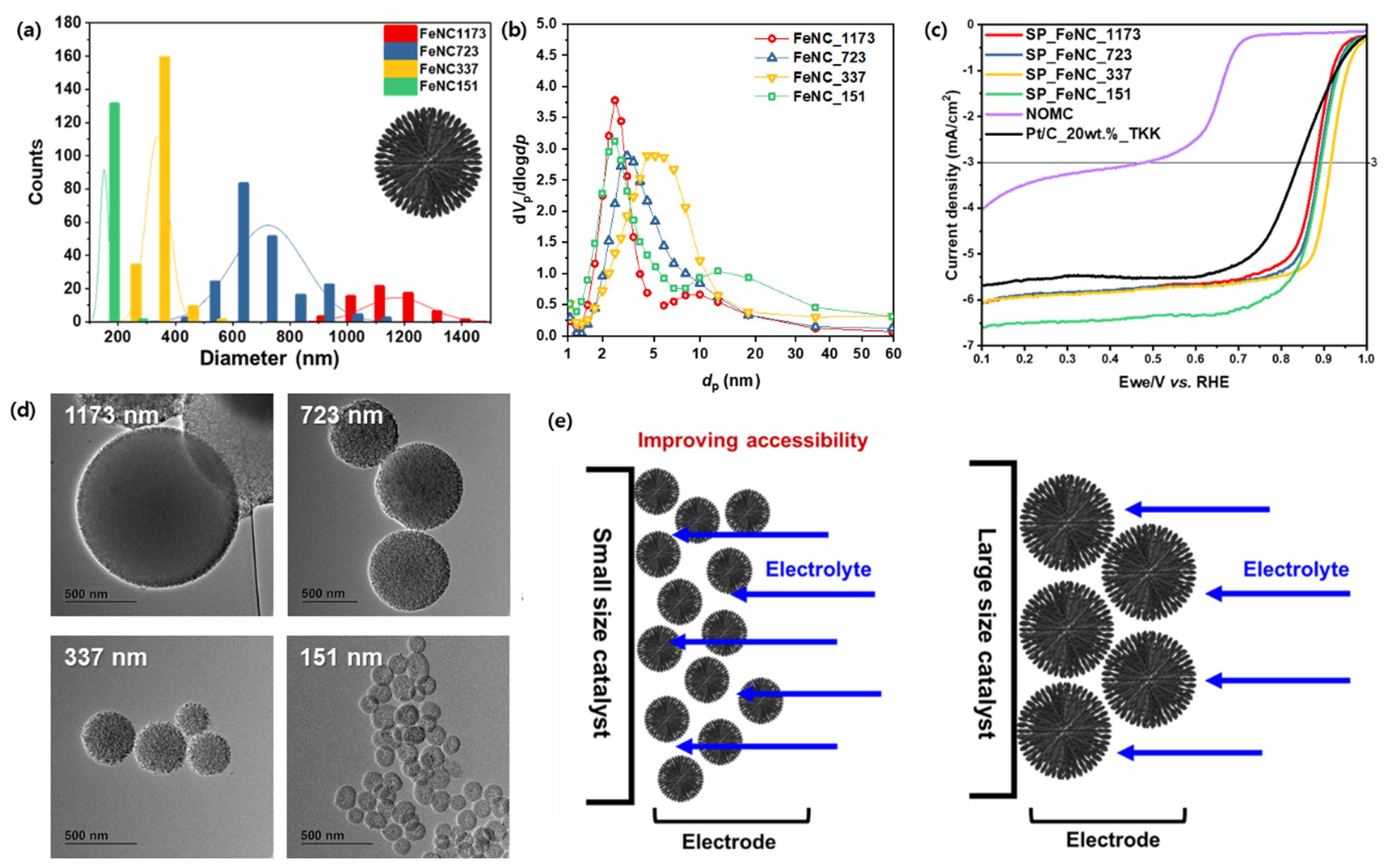
The particle size of MNC would affect the ORR activities in the aspect of the electrochemically active area. Lee et al. reported that mesoporous FeNC catalysts with different particle sizes would affect the electrochemically active area of catalysts [71]. Mesoporous FeNC catalysts were prepared by a hard-template method using mesoporous silica spheres varying in diameters as 1173, 723, 337, and 151 nm (Fig. 3a,b,d). Mesoporous silica spheres were prepared by modifying StÃķberâs method using n-hexa-decylamine as a soft template. The size of silica spheres was controlled by the added amount of ammonium hydroxide solution. Iron chloride and 1, 10-phenanthroline were infiltrated into the pores of silica templates and pyrolyzed at 900oC. The silica template prevented the agglomeration of iron nanoparticles during pyrolysis and led to the development of the mesopore structure of FeNC. It was concluded that the smaller particle size of the catalyst enabled enhanced electrolyte accessibility and led to improved ORR activities (Fig. 3c,e). It was maximized at the diameter of 337 nm with 0.918 V of half-wave potential and 5.881 mA cmâ2 of limiting current density. Agglomeration of metal particles at the diameter of 151 nm resulted in the hindrance of M-Nx site formation. It indicated that FeNC with smaller particle sizes would have a high electrolyte accessibility, nevertheless, it could be vulnerable to aggregation.
High active site density and porosity of the catalyst are essential for high ORR activity. Chen et al. reported high accessible active site density and hierarchically porous SA-Fe-NHPC by heat treating 2,6- diaminopyridine/ZnFe/SiO2 complex [72]. The high density of the active site was secured through two pyrolysis processes involving Fe precursor. Silica colloid contributed to mesopore formation, and Zn suppressed the formation of iron carbide. SA-Fe-NHPC created through this has a hierarchical pore structure and a high density of Fe-N4 sites (Fig. 4aâc). Comparing SA-Fe-NHPC to NP-Fe-NHPC without Zn, it had a higher porosity, and the effect of Zn on pore formation was clear (Fig. 4d,e). This showed high durability, maintaining performance at a high half-wave potential of 0.93 V and an accelerated stress test of 10,000 cycles (Fig. 4f,g).
2.3. Active site engineering of MNCs for facilitated ORR kinetics
What determines the kinetics of a catalyst is the adsorption energy between the active site and the reaction intermediate [73]. If the binding is too weak, the intermediate will be desorbed before the reaction proceeds. If the binding is too strong, the intermediate will not be desorbed and the active site will be inactive. Binding energy can be adjusted by adding electron donating or withdrawing chemical species to FeNC. High ORR activity can be achieved when the binding energy is at the proper range.
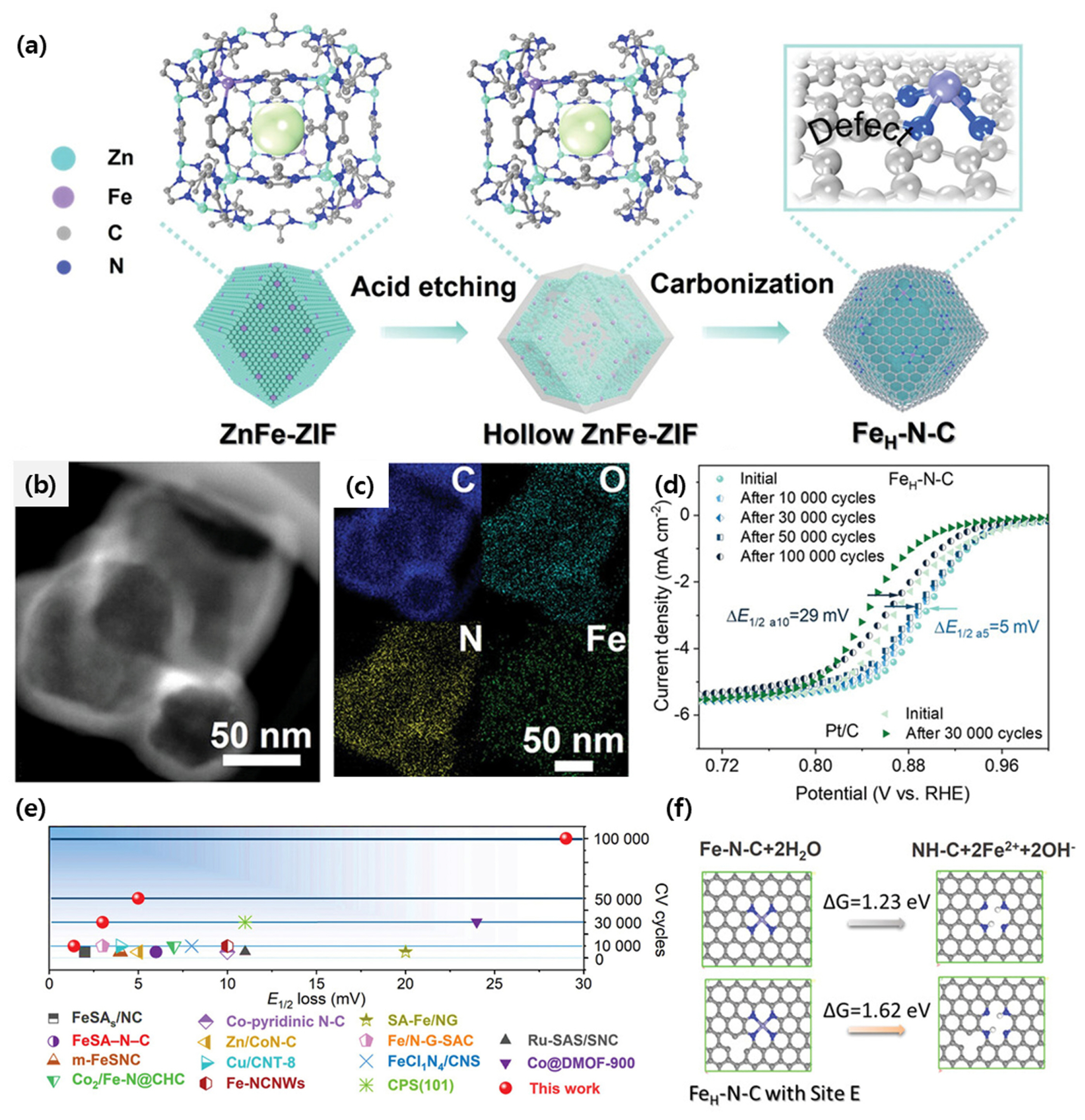
Tian et al. reported a FeH-N-C catalyst with carbon vacancy engineering with superior durability [74]. In synthesizing FeNC using ZIF-8, the zeolitic imidazole framework (ZIF) was etched with tannic acid to remove unstable chemical species, and it induced the formation of carbon defects during pyrolysis (Fig. 5a). The hollow structure of FeH-N-C, which was completed by heat treatment in an inert gas atmosphere (Fig. 5b,c). It showed high activity and durability, with the half wave potential decreasing by about 30 mV from 0.91 V to 0.88 V even after a durability test of 100,000 cycles in O2 purged 0.1 M KOH electrolyte solution, which was unprecedented durability for MNC catalysts (Fig. 5d,e). As a result of computations, high stability of the active site was expected because the Fe dissolution energy between Fe and N in Fe-N4 located at the defective carbon of FeH-N-C is higher than those of pristine FeNC (Fig. 5f).
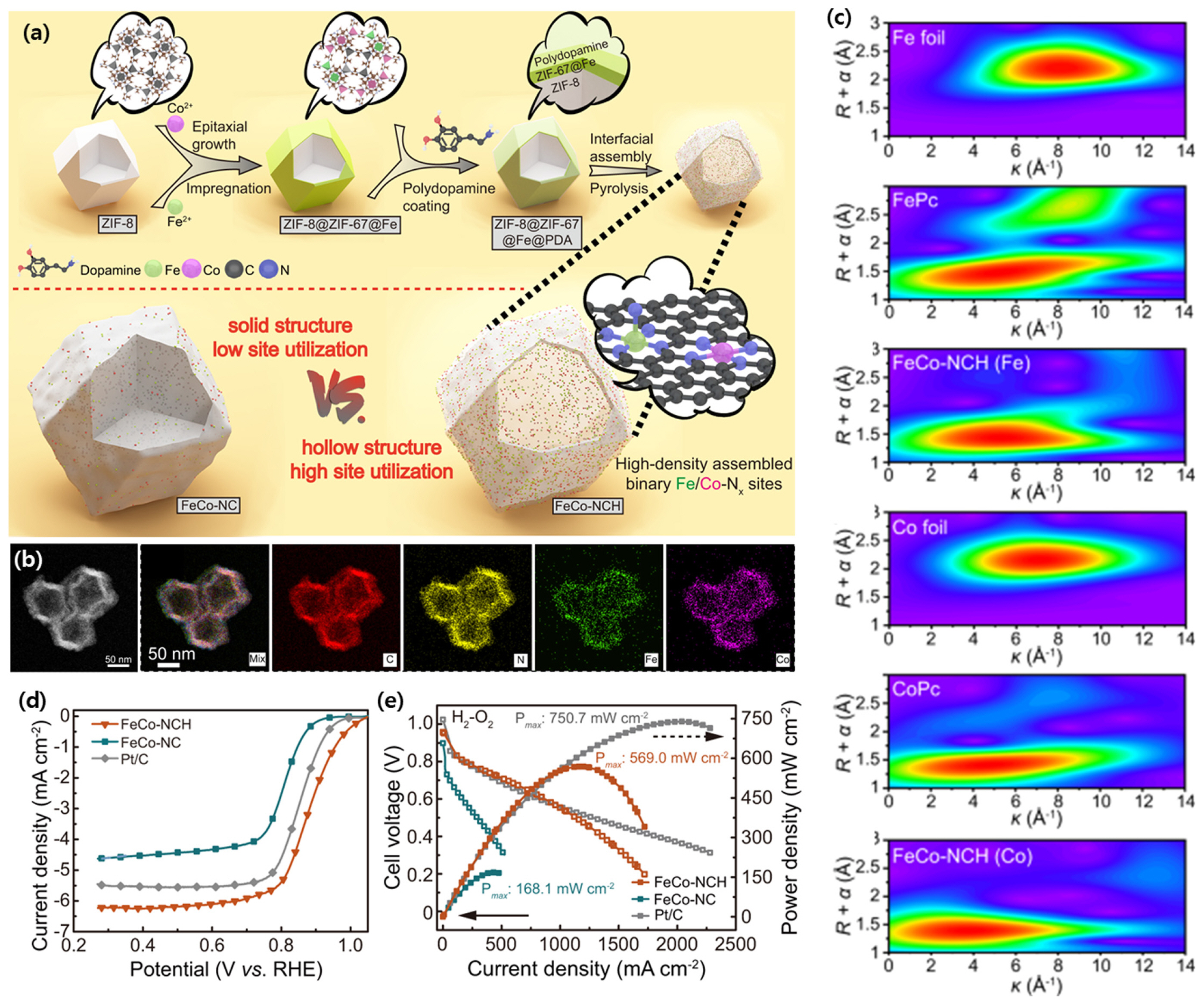
Jiang et al. formed binary single-atomic Fe/Co-Nx sites in a porous shell through interfacial assembly (FeCo-NCH) [60]. FeCo-NCH was prepared by pyrolyzing Fe and Co salts loaded ZIF-8 with poly-dopamine coating (Fig. 6a). As a result, FeCo-NCH was synthesized in the form of porous hollow particles containing C, N, Fe, and Co (Fig. 6b). Both Fe and Co have a coordination environment similar to metal phthalocyanine in a metal-N coordinated form rather than nanoparticles, and it was confirmed that they existed as the Fe-N5 and Co-N4 sites through EXAFS analysis (Fig. 6c). These active sites were highly accessible from oxygen and electrolyte and have a high half-wave potential of 0.89 V at 0.1 M KOH because the Fe/Co-Nx site has favorable electronic modulation for ORR (Fig. 6d). When applied as an AEMFC cathode, it showed a high peak power density of 750.7 mW cmâ2, showing the synergistic effect of a highly accessible active site with high ORR kinetics (Fig. 6e). Liu et al. reported that in the case of a highly coordinated structure in which the axial sulfur atom was located in the first shell and the phosphorus was located in the second shell, the adsorption energy of oxygen intermediates was weakened by down-shift of the d-band center [75]. The addition of the 4, 4â-sulfonyldiphenol, and phos-phonitrilic chloride trimer to the ZIF-8 synthesis procedure led to the introduction of S and P into the ZIF-8. The Fe-N4 site located in the hollow carbon nanocage containing N, P, and S showed high half-wave potentials of 0.912 and 0.814 V in alkaline and acidic media, respectively.
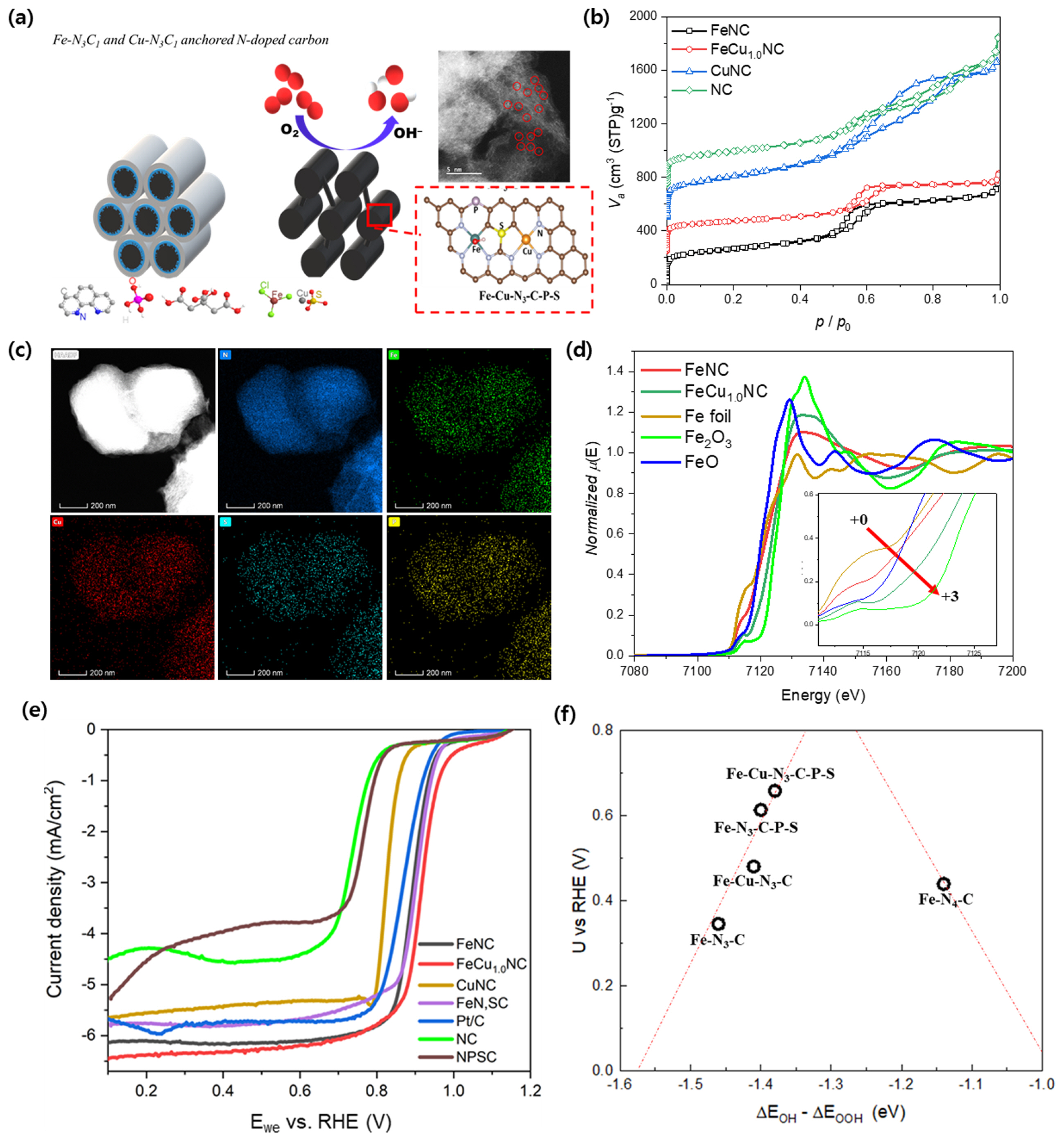
The electronic state of the active site can be adjusted by introducing other metals or heteroatoms. FeCuNC was synthesized using grain-shaped SBA-15, an ordered mesoporous silica template by carbonization of FeCl3, CuSO4, 1,10-phenanthroline, citric acid, and phosphoric acid impregnated SBA-15 (Fig. 7a) [61]. The use of the SBA-15 template was advantageous for mesopore structure and active site formation by limitation of the Fe agglomeration (Fig. 7b). Cu, S, and P shown in EDS mapping contributed to the positive shift of oxidation state of the active site despite being located above the second shell of the Fe-N3C1 site (Fig. 7c,d). The coordination environment of Cu was similar to that of Fe as Cu-N3C1, however, the shift of the oxidation state of Cu due to heteroatoms was negligible. Additionally, XPS and XAS revealed that S and P did not have a direct interaction with metals. Thus, it was concluded that heteroatom doping in the carbon matrix could shift the electronic state of Fe even located at the second or higher shell of the active metal. ORR activity was 0.92 V at â3 mA cmâ2 in half-cell (0.1 M KOH) and current density was 0.490 A cmâ2 at 0.6 V in AEMFC single cell (Fig. 7e). DFT calculations on catalyst model based on the results of XAS suggested that introduction of Cu, S, and P into FeNC improved ORR kinetics controlling the binding energy of OH* and OOH*, reaction intermediates (Fig. 7f).
3. Conclusion and Perspectives
This review summarized the development history of MNC catalysts, engineering of the active site (M-Nx moieties) for improved reaction kinetics, and approaches to improving mass transport ability through morphology control. In addition, the synthesis methodologies applied in recent studies, the characterization techniques for catalyst properties, and the resulting half-cell and single-cell performances for AEMFCs were summarized. Recent studies have shown that the development of MNCs with high performance and stability by overcoming sluggish ORR kinetics, satisfying two conditions. First, the modification of the electronic state of active sites through heteroatom doping, metal coordination change, and defect engineering to shift the position of the d-band, which led to improved reaction kinetics with suitable binding energy. The other is the morphology control of catalysts in the aspect of particle size or pore structure, which could easily expose the active site to reactants, resulting in a low mass transport resistance. Developed MNC catalysts would be expected to be applicable not only to fuel cells but also to other electrochemical devices (electrochemical CO2 reduction, metal-air batteries) that consider similar active sites.
MNC catalysts that had catalytic activities similar to or exceeding platinum in half-cells have been reported, however, they still show lower performance than platinum in single cells. This limitation is thought to be because most of the existing electrode structure research focuses on platinum catalysts, which indicates the lack of research on the design of electrode structures suitable for MNC. To implement a fuel cell with high performance using MNC cathodes, electrode structure design for MNC catalysts should be sufficiently considered in the aspect of the effect of surface properties of the catalyst, distribution of ionomers, and changes in electrode structure.