1. Introduction
As global awareness of environmental pollution caused by fossil fuels and the finiteness of resources has increased, interest in environmentally friendly renewable energy has risen sharply. In particular, fuel cell technologies are spotlighted due to their acceptable efficiency and sustainability [1]. They are environmental-friendly, noise-free, steady, and efficient when active catalysts are used on both electrodes [2]. There are several types of fuel cells, such as low temperature-operated fuel cell (direct methanol fuel cell (DMFC) and proton exchange membrane fuel cell (PEMFC)) and high temperature-operated fuel cell (solid oxide fuel cell (SOFC) and molten carbonate fuel cell (MCFC)) [3–5]. Low temperature-operated fuel cell facilitates devices and mobility things due to its unique operating temperature.
Nevertheless, there are some challenges for transportation because the fuel hydrogen is a gas fuel [6,7]. Direct liquid fuel cells with transport advantages have been focused on and studied, such as methanol, formic acid, hydrazine and so on [8–11]. Direct methanol fuel cell (DMFC) has been developed as a portable power source because of its excellent fuel availability, high energy density, and fuel abundance. Notwithstanding that DMFC shows pros in this issue, it is complicated beyond cons since methanol crossover is a serious problem [12–14]. In an acidic DMFC at low temperature, methanol fuel entering the anode passes through the electrolyte membrane. Also, the liquid fuel enters the cathode, reacting with the cathode catalyst. Therefore, methanol oxidation reaction (MOR) occurs at the cathode, resulting in a voltage drop [15–17].
It is highly required to progress methanol-tolerant Pt-based electrocatalysts with the prominent material to operate in low-temperature fuel cells with considerable and remarkable activity in ORR [18]. Many research groups have been developing to overcome this drawback; Pt nanoparticles covered by a thin carbon layer can be methanol tolerant catalysts. Wen et al. synthesized methanol-tolerant core-shell Pt/C nanoparticles whose mesoporous carbon shell protects Pt from methanol [18]. N-doped carbon layer plays a role in blocking methanol adsorption and enhancing ORR activity [19]. However, it is hard to control the thickness of the carbon layer in the synthesis process. The thick carbon layer can narrow the electrochemically active surface of Pt for ORR. For this reason, approaches using chemisorbed anions have also been suggested as methanol adsorption blockers such as cyanide radical [20] or Cl− [9]. Pt nanoparticles anchored to anion showed high methanol tolerant characteristics. It led to the improvement of DMFC performance by reducing the mixed potential [9].
Herein, we introduced the phosphate decorated platinum catalyst to diminish methanol adsorption ability as adsorption blockers synthesized by the modified polyol method. We confirmed that phosphate ion bonds to the carbon of Pt/C catalyst and the surrounded phosphate ion on Pt surface suppressed methanol oxidation reaction by blocking the methanol adsorption. Furthermore, the ORR activity retains regardless of these circumstances not to interrupt reactions. We demonstrated methanol tolerance of the catalyst with electrochemical analysis and DMFC single-cell analysis in various methanol concentrations.
2. Experimental
2.1. Synthesis of Pt/C decorated phosphate ion
Pt nanoparticles decorated with phosphate ion were synthesized by a similar procedure to our previous studies [21,22] based on a modified polyol method [23] using TRITONTM H-66 (DOW, K(CH3C6H6)PO4) which is an anionic surfactant by PO42−. We added TRITONTM H-66 into a mixed solvent of 132 mL of ethylene glycol (Sigma-Aldrich) and 68 mL of deionized (D.I.) water (Mili Q 18 Mohm). Using a bath sonicator, conductive carbon (300 mg, Vulcan XC-72R, CABOT) as carbon support for Pt nanoparticles was uniformly dispersed in the mixed solvent. Pt precursor solution was prepared by dissolving H2Pt-Cl6·xH2O (960 mg, Alfa Aesar) in D.I. water to make a 20 wt.% solution, then we diluted the Pt precursor solution with 20 mL of ethylene glycol. After two mixture solutions, we adjusted the pH of the mixture to 10 using 1 M NaOH solution (Sigma-Aldrich) and refluxed at 160°C for 3 hr. After reaching room temperature, we separated synthesized Pt/C catalyst powder from the emulsion by a centrifugal separator and filtered the catalyst powder several times. To eliminate moisture in the catalyst, it was dried in an oven at 60°C overnight and pyrolyzed in an atmosphere of 96% N2 + 4% H2 at 300°C for 1 hr. We denoted the synthesized Pt catalysts with TRITONTM H-66 as Pt/C-P. For comparison, we also synthesized a Pt catalyst without the surfactant, Pt/C [24].
2.2. Physico-chemical Analysis
The morphologies of synthesized Pt/C-P and Pt/C were observed with Field Emission Transmission Electron Microscope (FE-TEM, JEM-2100F, JEOL LTD). We also used energy-dispersive X-ray spectroscopy (EDX) for element distribution and atomic percentage of Pt, P, and C [24]. For the crystal structure of Pt, we carried out X-ray diffraction (XRD, Miniflex II, Rigaku) using Cu Kα as the X-ray anodization source (λ = 0.15141 nm). To confirm P species and Pt oxidation state in Pt/C-P, X-ray photon spectroscopy (XPS, AXIS-NOVA, Kratos Inc.) analysis was performed with Al Kα source of X-rays at Korea Basic Science Institute.
2.3. Electrochemical Analysis
Electrochemical analysis was proceeded by potentiostat/galvanostat (Biologic, VSP). We used three electrodes in a batch-type cell with a cooling jacket to keep the constant temperature (297 K). The working electrode was prepared by the drop-casting method on the glassy carbon of the rotating ring disk electrode (RRDE; area = 0.2475 cm2). Before the casting, the surface of glassy carbon was polished by three kinds of alumina powder. For catalyst ink, 10 mg of catalyst was dispersed in a mixture of 1.44 mL of D.I. water, 0.8 mL of 2-propanol (Junsei) and 10 μL of 10% Nafion resin dispersion solution (Sigma-Aldrich) by a bath sonicator. In 30 min, we dropped 9.9 μL of catalyst ink onto the glassy carbon and dried it in an oven for 10 min at 60°C, resulting in a Pt loading amount of 100 μg/cm2. Reference and counter electrodes were Ag/AgCl (in 3 M KCl) and Pt wire (Alfa Aesar), respectively. To check electrochemical active surface area (ECSA), we performed cyclic voltammetry (CV) in N2-saturated 0.1 M HClO4 (Sigma-Aldrich, assay, 70 %) from 1.05 to 0.0 VRHE at a scan rate of 20 mV/s. The activity of oxygen reduction reaction (ORR) was observed by linear sweep voltammetry (LSV) from 1.05 to 0.0 VRHE in O2-saturated 0.1 M HClO4 with a rotating speed of 1,600 rpm at a scan rate of 5 mV/s. For the CO stripping experiment, we purged CO gas in 0.1 M HClO4 and applied constant potential, 0.1 VRHE for 10 min to adsorb CO molecules on the catalyst surface. To eliminate the remaining CO molecules in the electrolyte, N2 gas was purged for 20 min at the open circuit potential. Then, CV was carried out to observe CO electro-oxidation peak in the potential range from 0.05 to 1.2 V at a scan rate of 20 mV/s. In terms of the electrocatalytic activity toward methanol oxidation, CV method was employed in N2-saturated 0.1 M HClO4 + 1, 10, 50, 100 mM methanol from 1.05 to 0.0 VRHE at a scan rate of 20 mV/s. To evaluate the ability of methanol tolerance of Pt/C-P and Pt/C, we employed two methods; LSV in O2-saturated 0.1 M HClO4 + 10, 50, 100 mM methanol and chronoamperometry (CP). For CP, we purged N2 gas in 0.1M HClO4 electrolyte for an initial 200 s and changed to O2 gas for 600 s. After the current density reached the limiting current, we added methanol into the electrolyte to make 0.1 M HClO4 + 100 mM methanol.
2.4. DMFC single cell test
To confirm the methanol tolerance by P-doped carbon layer in an actual fuel cell operation environment, we fabricated a membrane assembly electrode (MEA) with 9 cm2 active size and evaluated DMFC single cell performance using a fuel cell station (Scitech Korea, Inc.). Catalysts were Pt-Ru black (HISPEC, Alfa-aesar) and as-synthesized Pt/CP for anode and cathode, respectively, dispersed in the mixture of 1-propanol (Junsei), 2-propanol, D.I. water and 10 wt.% Nafion resin solution by a probe sonicator. The catalyst inks were then loaded on the gas diffusion layer (GDL, AvCarb MGL190 for anode and SGL 39BB for cathode) via hand spray. The target loading amount was 3.0 mgPtRu/cm2 and 1.0 mgPt/cm2 for anode and cathode, respectively. Two as-prepared electrodes were assembled with a proton exchange membrane (Nafion® 115, Dupont) and pressed for 5 min at 140°C. The resulting MEAs were placed between two graphite bipolar plates with a single serpentine flow pattern. The DMFC single cell was operated at 80°C of cell temperature with various concentrations (0.5, 0.75, 1 and 3 M) of methanol aqueous solution as the anode fuel and O2 gas for the cathode. The flow rates of methanol solution and O2 gas were 5 ml/min and 250 sccm. In the I–V curve measurement, each point was produced every 10 mA/cm2 of current density and maintained for 20 s at the current density.
3. Results and Discussion
We carried out HR-TEM analysis to check the morphology change of Pt/C-P by P-containing surfactant. Surfactant can affect the change in crystal shape, size, and degree of dispersion of nanoparticle. Fig. 1(a) and Fig. S1 indicate Pt nanoparticles of both Pt/C and Pt/C-P were successfully loaded on the carbon support. The P-containing surfactant did not modify the crystal change of Pt nanoparticle of Pt/CP compared to Pt/C. However, there were a couple of aggregated Pt particles in pristine Pt/C. Mean diameter of Pt nanoparticles of Pt/C (5.5 ± 1.8 nm) was slightly larger than that of Pt/C-P (5.0 ± 1.1 nm). Amphiphilic P-containing surfactant, consisting of benzyl and phosphate ion, could help the uniform nucleation of Pt4+ ion via electrostatic force during the polyol process [25]. Furthermore, we observed thin layers on Pt nanoparticles of Pt/C-P in Fig. 1(b). 0.5% of the atomic percentage of P atoms remained on Pt/C-P catalyst as EDS result in Fig. 1(c).
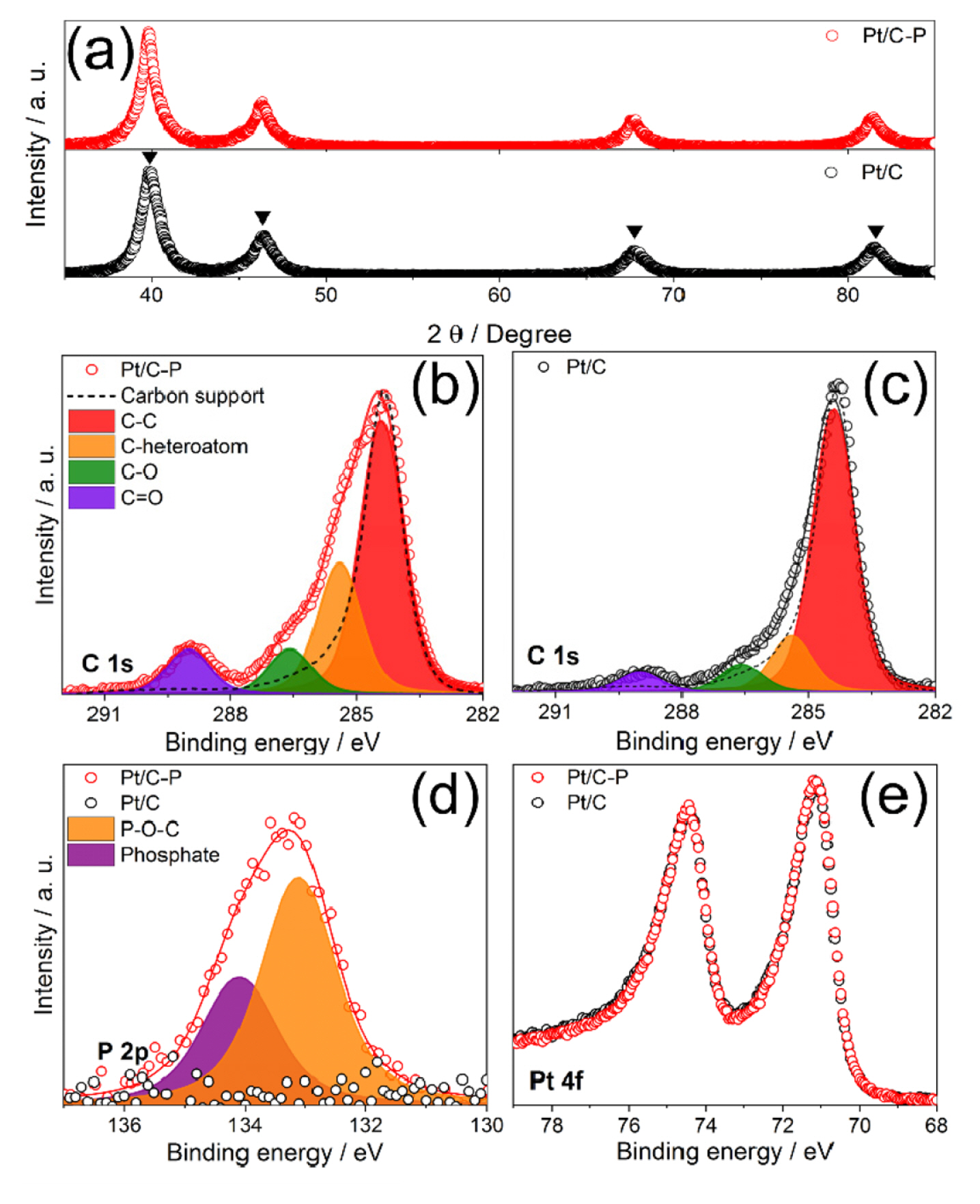
To clarify the crystallinity of Pt nanoparticle which can contribute the electrocatalytic activity toward ORR and methanol oxidation [22,26], XRD analysis was performed. Both Pt/C and Pt/C-P showed identical XRD patterns indicating facial centered crystal structure (FCC) of Pt [27] having four 2θ peak at 39, 47, 67, and 81° according to Pt (111), (200), (220), and (311), respectively (Fig. 2(a)). Corresponding to HR-TEM results, the (111) crystal domain size calculated by Scherrer’s of Pt/C and Pt/C-P were 6.3 and 5.9 nm. To figure out the P-related species on the Pt/C-P, XPS spectra of Pt/C-P in C 1s region were compared with Pt/C and bare carbon support. As shown in Fig. 2(b,c), width of spectra of Pt/C and Pt/C-P were wider than bare carbon support, indicating that some functional group such as C=O and C-heteroatom could be produced on carbon support during the Pt nanoparticle synthesis process. To find out the functional group, the XPS spectra were deconvoluted with four peaks: C-C or C-H (~284.6 eV), C-heteroatom (~285.8 eV), C-OH, alcohol (~287.2 eV), and C=O (~289.2 eV) [28]. Note that the intensities of C=O and C-heteroatom peaks were highly increased in only Pt/C-P. As shown in Fig. 2(d), only Pt/C-P had P 2p peak, which was deconvoluted with P-O-C and phosphate at 133.1 and 134.0 eV, respectively [29]. P atoms in Pt/C-P existed as P-O-C species, which could be found in P-doped carbons from several literatures [29–32], thus the enhanced C-heteroatom peak in C 1s spectrum of Pt/C-P resulted from the formation of linkage of P atom and carbon support. Considering C-O and C=O peak in XPS spectra in C 1s of Pt/C-P, some phosphate ions remained on the surface of Pt/C-P and directly bonded to C of the carbon support. On the other hand, there was no change in Pt oxidation state between Pt/C and Pt/C-P, indicating phosphate ions around Pt nanoparticles could not modify the d-band structure of Pt (Fig. 2(e)).
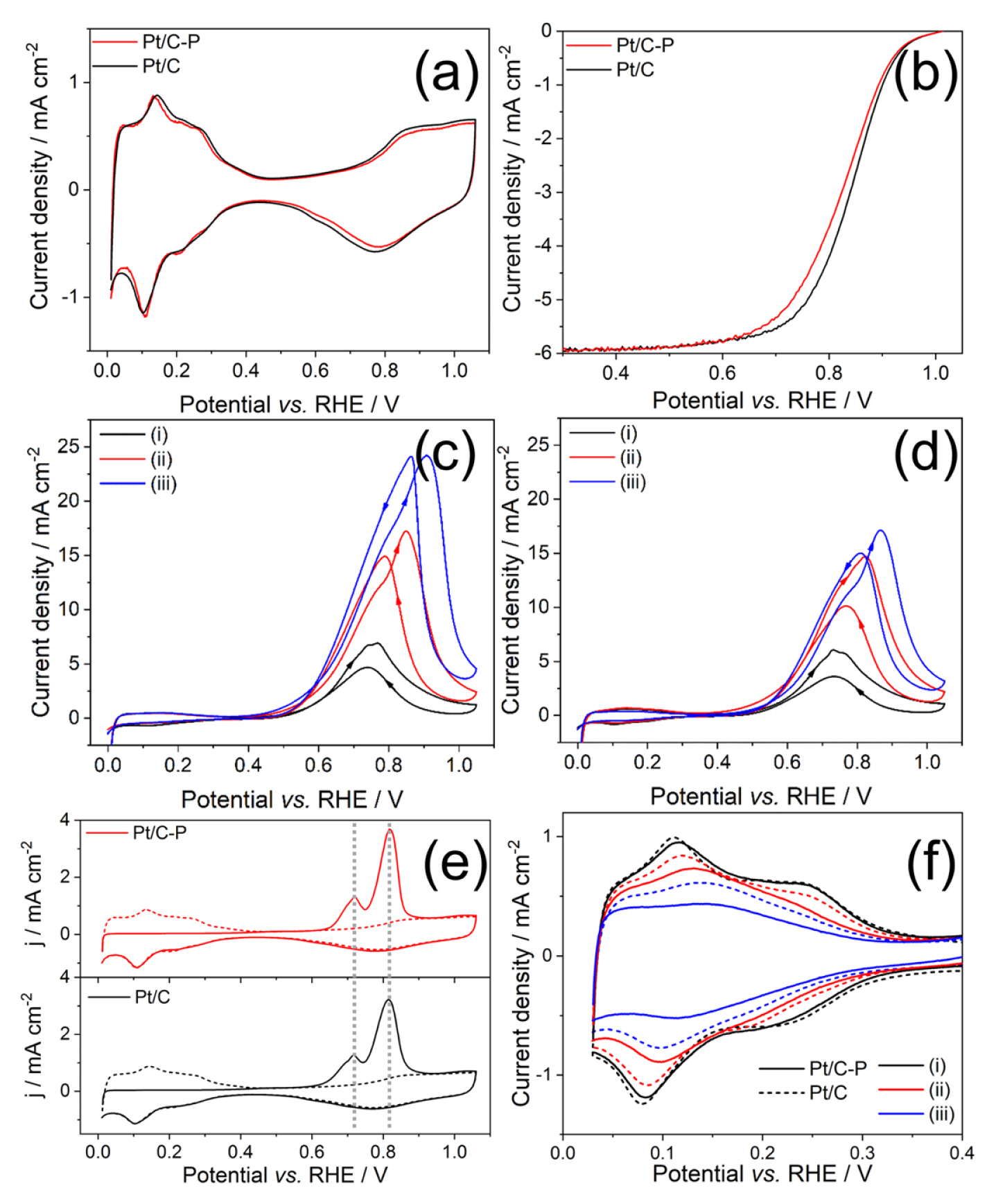
Phosphate ions on the catalyst surface can act as poisoning species which block the reactant and lower the ECSA. To compare ECSA, we carried out CV analysis in N2-saturated 0.1 M HClO4 as shown in Fig. 3(a) and calculated ECSA with the oxidation peak from 0.0 to 0.45 VRHE, which is responsible to hydrogen desorption on Pt surface [33,34]. ECSA of Pt/C (40.29 m2/g) was similar to Pt/C-P (40.55 m2/g), while the ORR activity of Pt/C-P was slightly lower than that of Pt/C (Fig. 3(b)). The half-wave potential, known as the ORR activity indicator [35], of Pt/C-P, was negatively shifted around 25 mV since phosphate ion on Pt(111) can block the Pt active site from the adsorption of O2 for ORR [36,37]. Despite this adverse effect, the amount of phosphate ion was only around ~0.5%, thus not enough to cover the full Pt site for ORR. Methanol-tolerant Pt catalyst should have not only good ORR activity but also low activity toward methanol oxidation. We also compared the CVs for methanol oxidation of Pt/C and Pt/C-P in N2-saturated 0.1 M HClO4 with various concentrations of methanol. (Fig. 3(c,d)) As methanol concentration increased from 10 to 100 mM methanol, the peak oxidation current densities of both forward direction (If) and backward peak (Ib) monotonically increased in Pt/C and Pt/C-P catalyst. Compared to Pt/C, Pt/C-P showed less activity toward methanol oxidation (If and Ib) and higher the current density peak ratio of If to Ib (If/Ib) every methanol concentration (Table 1). For several decades, numerous researchers reported the If/Ib is related with the degree of CO tolerance [38–40], thus a high If/Ib has been regarded as a crucial parameter to define outstanding methanol oxidation catalyst. However, other researchers suggested that Ib is attributed to the oxidation of freshly chemisorbed methanol on the free Pt surface, not residual CO on the catalyst since residual CO intermediates are eliminated during the forward scan [41,42].
Using electrochemical impedance spectroscopy, it was also revealed that the origin of the hysteresis between If and Ib results from the Pt surface coverage effect, which leads the switch of rate-determining step (RDS) from OH adsorption on CO adsorbed Pt surface by water dissociation to methanol dehydration (adsorption) on free Pt surface. Therefore, they suggested If/Ib is responsible to the degree of oxophilicity [42]. Increased If/Ib in Pt/C-P compared to Pt/C could be interpreted as high oxophilicity of Pt/C-P due to phosphate ion bonded to carbon, implying Pt surfaces are covered by P-oxygenated species. It leads to degenerate Ib peak by hindering methanol adsorption on free Pt surface. Not only Ib peak, If peaks of Pt/C-P were reduced, thus we tried to understand the origin of If reduction based on the proposed methanol oxidation mechanism. In contrast with Ib, which is attributed from direct methanol oxidation on free Pt surface, If involves CO formation from methanol adsorption via C-H bond breaking (dehydration) at low potential range (< 0.05 VRHE) in Eq. (1) [43] and CO oxidation with OH in Eq. (3), produced by water dissociation on Pt in Eq. (2), from 0.6 VRHE consecutively [42].
This mechanism is based on the Langmuir-Hinshelwood mechanism [44], and it is known that Eq. (3) is suppressed due to sluggish Eq. (2) as RDS. Therefore, we studied the CO oxidation behavior by CO stripping experimental as shown in Fig. 3(e), indicating the same trends in Pt/C and Pt/C-P. There was no shift of CO oxidation peak between Pt/C and Pt/C-P. It means phosphate on Pt surface in Pt/C-P could affect CO formation via methanol adsorption rather than CO oxidation accompanying water dissociation. To compare methanol dehydration reaction, which starts at 0.05 VRHE, we swept the potential from 0.03 VRHE to exclude hydrogen oxidation/evolution reaction in various methanol concentrations as shown in Fig. 3(f). We could not directly observe the methanol adsorption peak opposite to the previous study [45]; however, we noticed the suppression of both adsorption and desorption peak of under-potential deposited hydrogen (Hupd) due to methanol adsorption on Pt surface. Pt/C-P showed relatively small suppression of Hupd peaks, indicating it is hard to absorb methanol on Pt/C-P. It could allow reducing the If peak in Pt/C-P.
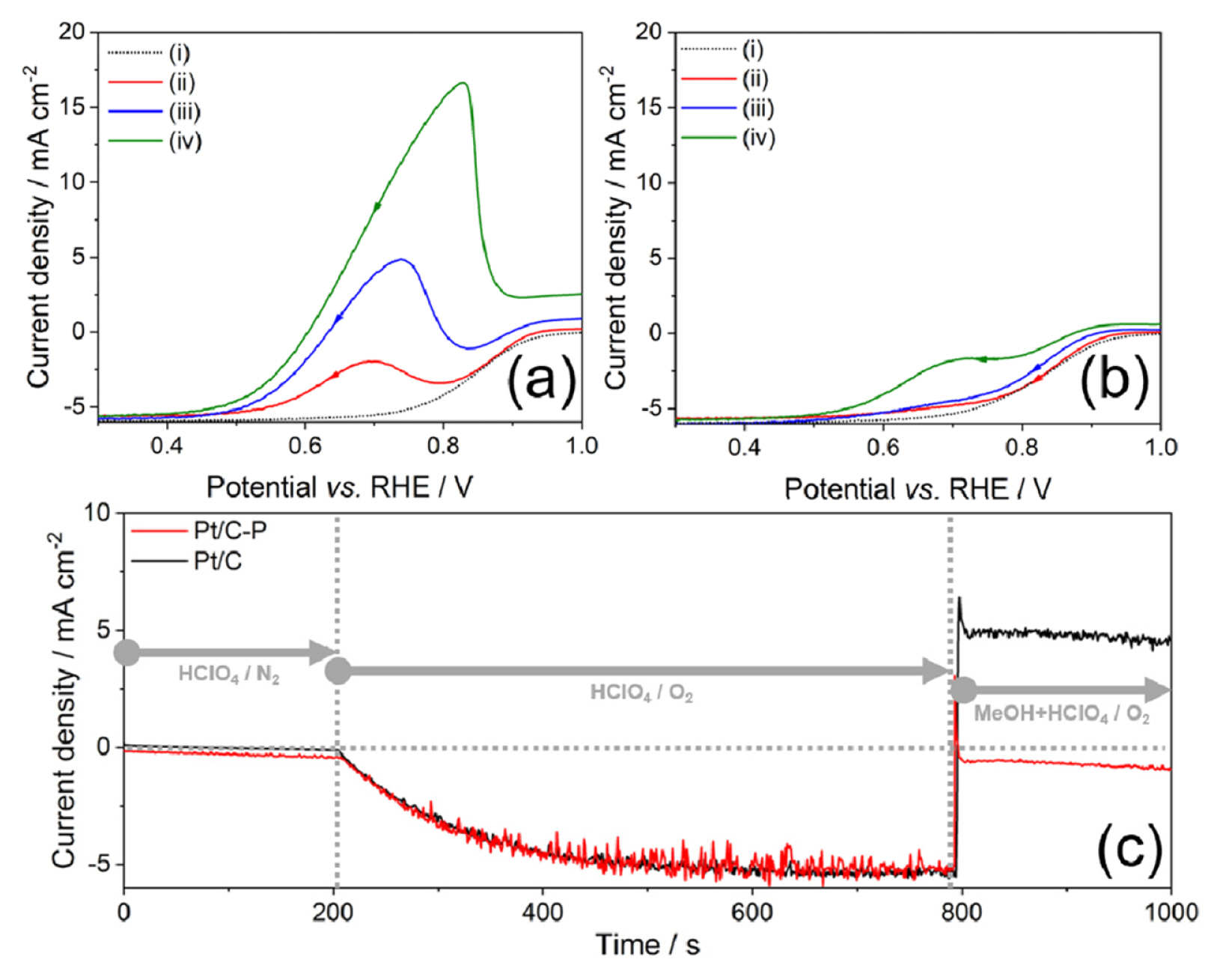
To evaluate the methanol-tolerant ORR performance, we carried out LSV in O2-saturated 0.1 M HClO4 with various concentrations (10, 50, 100 mM) of methanol. It could give an insight into DMFC performance, which is easily damaged by permeated methanol to the cathode through proton exchange membrane. Fig. 4(a) shows large oxidation peaks from methanol oxidation despite the oxygen (O2) atmosphere, indicating methanol oxidation is more favorable on Pt/C than ORR. On the other hand, Pt/C-P had high methanol-tolerant ORR performance by tracing the LSV in 0 mM methanol + 0.1 M HClO4 electrolyte in Fig. 4(b). Considering low Ib and high If/Ib in Fig. 3(d), Phosphate ion linked to Pt surface could block the adsorption of methanol, but permeate O2 molecule selectively and allow to be reduced. We speculate that this discrepancy in the adsorption ability is related to the molecular size of methanol and O2. In order to determine the degree of methanol oxidation reaction against ORR, the current-time response was examined using CA at 0.7 VRHE where If appeared. Current responses of Pt/C and Pt/C-P were similar in both N2 and O2-saturated 0.1 M HClO4 electrolyte, as shown in Fig. 4(c). However, as soon as methanol was injected into the electrolyte, the current density of Pt/C was suddenly switched to oxidation current due to vigorous methanol oxidation reaction. In contrast, that of methanol tolerant Pt/C-P still stayed in negative current density. We also observed the same tendencies of Pt/C-P and Pt/C at other CA potentials from 0.65 to 0.85 VRHE (Fig. S3).
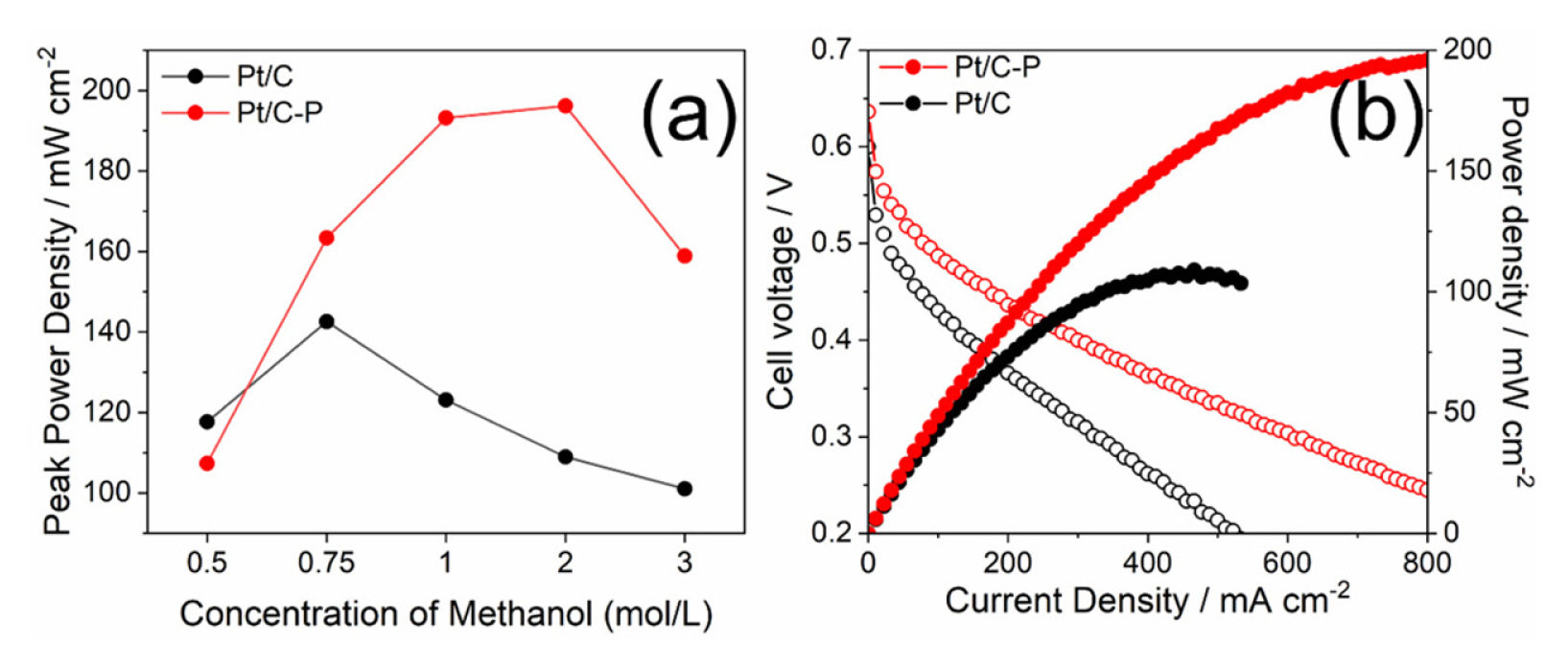
We finally performed DMFC single-cell operation via I–V curve measurement to evaluate the methanol-tolerant ability of Pt/C-P and Pt/C as cathode catalyst with 0.5, 0.75, 1.0, 2.0 and 3.0 M methanol. As increasing methanol concentration in anode side, the methanol flux by methanol crossover from anode to cathode is greater [15]. Therefore, peak power density (PPD) of Pt/C at the lowest methanol concentration (0.5 M) was slightly higher than that of Pt/C-P with lower ORR activity (Fig. 5(a)). At higher methanol concentration than 0.5 M, Pt/C-P showed significantly higher PPD than Pt/C due to its methanol-tolerant property by phosphate. Not only PPD, open circuit voltage (OCV) could be an indicator describing the methanol-tolerant property in DMFC system. In Table 2, as increasing the methanol concentration, OCVs of both Pt/C and Pt/C-P were gradually decreased from 0.69 to 0.61 V due to the methanol crossover. At 0.5 M, Pt/C showed higher OCV than Pt/C-P since the activity loss by ORR activity was much crucial in OCV decision rather than iR drop by methanol influence at low methanol concentration. Except for 0.5 M, Pt/C-P showed higher OCV than Pt/C. At 1.0 M methanol concentration, Pt/C-P obtained superb DMFC performance with 198 mW/cm2 of PPD at 0.27 V (Fig. 5(b)). The PPD of Pt/C-P showed comparable performances with previous literature (Table S1). The same tendency was observed with air as cathode gas (Fig. S5).
4. Conclusions
Phosphate-decorated Pt nanoparticles on carbon support (Pt/C-P) for methanol-tolerant cathode catalyst were synthesized by a modified polyol method using amphiphilic P-containing surfactant. The linkage of phosphate ions and carbon around the Pt surface was formed with modification of the carbon surface; meanwhile, Pt surface structure is not entirely changed. Phosphate ion did not affect the activity of ORR; however, the modified formation degenerates methanol oxidation activity by blocking methanol from adsorption on Pt surface in the cathode. As a result, Pt/C-P became resistant to methanol, maintaining activity toward ORR. Pt/C-P in the cathode of DMFC single cell showed much higher performance than Pt/C catalyst due to its accomplished selectivity toward ORR. This work suggests a methodology to resist the methanol crossover for high DMFC performance and develop the Pt-based catalysts linked to heteroatom-based anion.